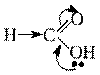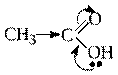Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Классификация органических соединенийСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Центрифуги
Перед центрифугированием центрифужные пробирки уравновешивают и располагают в центрифуге симметрично. Необходимо, чтобы центрифужная камера была закрыта крышкой. Во время работы центрифуги запрещается открывать крышку камеры. После отключения центрифуги необходимо дать возможность ротору остановиться, запрещается тормозить ротор рукой.
Автоматические пипетки
Принципы дозировки: –– поворотным движением надеть наконечник на шток пипетки, — держа пипетку в вертикальном положении нажать кнопку до первого упора (фаза А), — погрузить наконечник в жидкость на глубину 3 до5 мм(фаза В), — набрать жидкость в наконечник медленно отпуская кнопку. Немного подождать и вынуть наконечник из жидкости (фаза С), — прикоснуть наконечником к внутренней стенке намеченного сосуда и опорожнить наконечник, плавно нажимая кнопку до первого упора с такой же скоростью как при взятии пробы (фаза D), — подождать около 1 секунды, — нажимая кнопку пипетки до второго упора, удалить остатки жидкости и вынуть пипетку, скользя наконечником по внутренней стенке сосуда (фаза Е), –– после снятия наконечника, пипетка готова к повторению цикла работы.
Запрещается набирать жидкость без предварительно наложенного наконечника. Нельзя погружать наконечник в жидкость глубже5 мм. Нельзя допустить, чтобы большой палец соскользнул с кнопки во время наполнения наконечника. Нельзя поворачивать пипетки, когда наконечник наполнен жидкостью или мокрый. Перемещение кнопки пипетки при наполнении и опорожнении наконечника должно осуществляться плавно и. медленно. Во время дозировки жидкостей, смачивающих стенки наконечника (например: сыворотки, белка, органических растворителей), рекомендуется предварительно прополоскать несколько раз наконечник измеряемой жидкостью: в наконечник, наложенный на пипетку, нужно набрать и вылить из него жидкость, повторяя несколько раз эту операцию. При работе с жидкостью, имеющей температуру, отличающуюся от температуры окружающей среды более 5 °С также рекомендуется многократно прополоскать наконечники. После окончания работы следует поместить пипетку в штативе.
Вопросы для проверки полученных знаний 1. ОБЩАЯ ХАРАКТЕРИСТИКА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ.
1.1 Основные понятия и классификация органических соединений
Органические соединения — класс химических соединений, в состав которых входит углерод (за исключением оксидов углерода, карбидов, угольной кислоты, карбонатовицианидов). Название «органические соединения» появилосьнараннейстадииразвитияхимиииговоритсамозасебя–ученыетойэпохисчитали,чтоживыесуществасостоятизособых органических соединений. Структурная изомерия Количество изомеров С-скелета очень быстро возрастает с увеличением числа С-атомов:
Изомерия положения
Таутомерия «таутос» - тот же самый, «мерос» - доля, часть (греч.). Таутомерия – явление динамического обратимого превращения изомеров, протекающее с разрывом и образованием связей и сопровождающееся перемещением атомов (чаще всего протона) и реже групп атомов. Изомерные формы – таутомеры. В отличие от структурных изомеров, таутомеры, как правило, не могут существовать отдельно друг от друга. Независимое их получение невозможно. Основным признаком таутомерных веществ является их двойственное реагирование – способность к образованию двух рядов производных как результат раздельного и самостоятельного реагирования двух находящихся в равновесии изомерных форм одного соединения. Виды таутомерии
Геометрическая изомерия - разновидность стереоизомерии, которая определяется различием пространственного расположения в молекулах пары заместителей относительно плоскости двойной связи или цикла. Обусловлена тем, что в молекулах этих веществ свободное вращение атомов вокруг σ-связей (циклоалканы) и относительно π-связей (алкены) оказывается невозможным.
Z,E-номенклатура (для три- и тетразамещенных алкенов). Конфигурацию изомера определяют по относительному расположению старших заместителей. По одну сторону плоскости – Z-изомер; по разные – Е-изомер. В основе определения старшинства находится атомный номер элемента. В случае одинаковых атомов старшинство группы определяет «вторая оболочка» атомов: -СН3< -СН2СН3< -СН(СН3)2< -СН2NН2< -CH2OH< -CH2F В случаях групп с различным типом связи старшинство увеличивается в рядах: -CH2OH < -COH < COR < COOH -CH2NH2< -CH=NH < -CN
Е-изомер Z-изомер Вследствие того, что расстояния между заместителями в молекулах изомеров различны, последние существенно различаются своими химическими и физическими свойствами. Они могут быть разделены и существовать индивидуально.
Переход одного изомера в другой – изомеризация протекает обычно при нагревании или облучении.
Конформационная изомерия - разновидность стереоизомерии, которая определяется различием пространственного расположения в молекулах заместителей, возникающее в результате свободного вращения вокруг σ-связей. Такие изомеры различаются между собой стабильностью. Более стабильные конформации, которые фиксируются физико-химическими методами, называются конформерами.
Изображение конформеров – проекции Ньюмена:
Чем больше сила взаимного отталкивания атомов водорода, тем выше энергия системы; поэтому заторможенной конформации будет соответствовать минимум потенциальной энергии молекулы. Принимая различные конформации, молекулы остаются химически однородными; конформации не являются типичными изомерами. Однако, в некоторых случаях (при тесной упаковке молекул), можно разделить различные формы. Конформации биоорганических молекул (ферменты, витамины, белки, нуклеиновые кислоты) играют определяющую роль в проявлении последними биологической активности. Коформации в ряду циклических углеводородов:
Конфигурационная изомерия Объёмные структуры циклических соединений содержат разные по природе положения заместителей:
Оптическая изомерия Некоторые органические соединения являются оптически активными. Они способны изменять плоскость поляризации света при прохождении его через образец вещества (1815 г. Ж. Био). Свет – электромагнитные волны, колебания которых перпендикулярны направлению их распространения. В естественном (солнечном) свете эти колебания происходят в различных плоскостях.
Оптически активные соединения поворачивают плоскость поляризации на определённый угол вправо (правовращающие) или влево (левовращающие). Изомеры, вращающие плоскость поляризации в разные стороны, но на один и тот же угол – антиподы (энантиомеры). Рацемическая смесь (рацемат) – смесь, состоящая из равных количеств лево- и правовращающих изомеров. Рацемат оптически не активен. Оптическая активность характерна для соединений содержащих
Изображение энантиомеров
Для того, чтобы связать строение с вращением, было предложено выбрать соединение-стандарт и сравнить с ним все другие соединения, содержащие хиральный центр. В качестве стандарта был выбран
R,S- номенклатура Для отнесения стереоизомера необходимо определить в нем старшинство заместителей (порядковый номер элемента – как в случае Z,E-изомерии). Взгляд наблюдателя направляется по оси С-младший заместитель (Н). После такой ориентации смотрят, как три заместителя располагаются в ряд в направлении от старшего к младшему. В случаеR-конфигурации этот порядок соответствует направлению движения по часовой стрелке, в случаеS-конфигурации – против часовой стрелки.
Если молекуле имеется несколько хиральных центров, то число изомеров возрастает и равняется 2n, где n - число хиральных центров.
В отличие от структурных изомеров энантиомеры идентичны один другому в большинстве своих свойств. Они отличаются только по своему взаимодействию с плоскополяризованным светом и взаимодействием с веществами, которые также являются хиральными. В организме реакции протекают с участием биокатализаторов - ферментов. Ферменты построены из хиральных молекул α-аминокислот. Поэтому они играют роль хиральных реагентов, чувствительных к хиральности взаимодействующих с ними субстратов (стереоспецифичность биохимических процессов). Это приводит к тому, что хиральные природные соединения представлены, как правило, лишь одной стереоизомерной формой (D-углеводы, L-аминокислоты). Стереоспецифичность лежит в основе проявления биологического действия одним из энантиомеров, в то время, как другой изомер может быть неактивным, а иногда оказывать иное или даже противоположное действие.
1.3 Химическая связь в органических соединениях
При образовании химической связи выделяется энергия, поэтому появление двух новых валентных возможностей приводит к выделению дополнительной энергии (1053,4 кДж/моль), которая превосходит энергию, затраченную на распаривание 2s электронов (401 кДж/моль). Различные по форме орбитали (s,p) при образовании связи смешиваются, давая новые равноценные гибридизованные орбитали (теория гибридизации, Л.Полинг, Д.Слэтер, 1928-1931 гг.). Понятие гибридизации относится только к молекулам, но не к атомам, и в гибридизацию вступают только орбитали, а не электроны на них. В отличие от негибридизованных s- и p- орбиталей гибридная орбиталь полярна (электронная плотность смещена) и способна образовывать более прочные связи.
Виды химической связи Ионная связь -возникает в случае полной отдачи электронов одними атомами и приобретением их другими. При этом атомы превращаются в ионы. Ковалентная связь - образуется путем обобществления электронов. Связывание атомов в молекуле осуществляется электронной парой, принадлежащей одновременно двум атомам. Обобществление электронов возможно двумя способами: 1) коллигация (обменный механизм); 2) координация (донорно-акцепторный механизм). Существует два типа ковалентной связи: σ (сигма)- и π (пи)- связи.
σ-связью называется одинарная ковалентная связь, образованная при перекрывании атомных орбиталей по прямой (оси), соединяющей ядра двух связанных атомов с максимумом перекрывания на этой прямой. π-связью называется связь, образованная при боковом перекрывании негибридизованных pz -атомных орбиталей с максимумом перекрывания по обе стороны от прямой, соединяющих ядра атомов. Количественные характеристики ковалентной связи 1. Энергия связи – это энергия, выделяющаяся при образовании связи или необходимая для её разрыва. 2. Длина связи – это расстояние между центрами связанных атомов. 3. Полярность связи – неравномерность распределения электронной плотности. 4. Поляризуемость связи – смещение электронов связи под влиянием внешнего электрического поля, в том числе и другой реагирующей частицы. Межмолекулярные взаимодействия
Органические кислоты В органических соединениях в зависимости от природы элемента, с которым связан Н+, различают следующие кислоты: ОН – кислоты (карбоновые кислоты, фенолы, спирты) СН – кислоты (углеводороды и их производные) NH – кислоты (амины, амиды, имиды) SH – кислоты (тиолы). Кислотным центром называется элемент и связанный с ним атом водорода. Сила кислоты будет зависеть от стабильности аниона, т.е. от сопряженного основания, которое образуется при отрыве Н+от молекулы. Чем стабильнее анион, тем выше кислотность данного соединения. Стабильность аниона зависит от ряда факторов, которые способствуют делокализации заряда. Чем выше делокализация заряда, тем устойчивее анион, тем сильнее кислотные свойства. Факторы, оказывающие влияние на степень делокализации: 1. Природа гетероатома в кислотном центре 2. Электронные эффекты атомов углеводородных радикалов и их заместителей 3. Способность анионов к сольватации. 1. Зависимость кислотности от гетероатома. Под природой гетероатома понимают его электроотрицательность (Э.О.) и поляризуемость. Чем больше (Э.О.) тем легче осуществляется гетеролитический разрыв в молекуле. В периодах слева направо с ростом заряда ядра растет (Э.О), т.е. способность элементов удерживать отрицательный заряд. В результате смещения электронной плотности связь между атомами поляризуется. Чем больше электронов и чем больше радиус атома, тем дальше электроны внешнего энергетического уровня расположены от ядра, тем выше поляризуемость и выше кислотность. Пример: СН- NH-OH-SH-
С, N,О – элементы одного периода. Э.О. по периоду растет, кислотность увеличивается. В этом случае поляризуемость влиять на кислотность не будет. Поляризуемость атомов в периоде изменяется незначительно, поэтому главным фактором определяющим кислотность является Э.О. Теперь рассмотрим ОН- SH-
О, S– находятся в одной группе, радиус в группе сверху вниз увеличивается, следовательно растет и поляризуемость атома, что ведет к увеличению кислотности. УSрадиус атома больше, чем у О, поэтому тиолы проявляют более сильные кислотные свойства по сравнению со спиртами. Сравнить три соединения: этанол, этантиол и аминоаэтанол: Н3С – СН2– ОН, Н3С – СН2– SH и Н3С – СН2– NH2 1. Сравним по радикалу – они одинаковые; 2. По природе гетероатома в функциональной группе: Sи О находятся в одной группе, но уSрадиус атома больше, поляризуемость выше, следовательно этантиол обладает более сильными кислотными свойствами 3. Теперь сравним О и N. О обладает более высокой Э.О., следовательно кислотность у спиртов будет выше. 2. Влияние углеводородного радикала и присутствующих в нем заместителей Необходимо обратить внимание студентов, что сравниваемые соединения должны иметь одинаковый кислотный центр и один растворитель. Электроноакцепторные (Э.А.) заместители способствуют делокализации электронной плотности, что ведёт к стабильности аниона и соответственно увеличению кислотности. Электронодонорные (Э.Д.) заместители наоборот способствуют концентрации электронной плотности в кислотном центре, что ведет к понижению кислотности и увеличению основности. Например: одноатомные спирты проявляют более слабые кислотные свойства по сравнению с фенолами. Пример: Н3С → СН2→ ОН 1. Кислотный центр один и тот же 2. Растворитель один и тот же В одноатомных спиртах электронная плотность смещается от углеводородного радикала к группе ОН, т.е. радикал проявляет +Iэффект, тогда на группе ОН сосредотачивается большое количество электронной плотности в результате чего Н+ более прочно связан с О и разрыв связи О-Н происходит трудно, поэтому одноатомные спирты проявляют слабые кислотные свойства. У фенола наоборот бензольное кольцо является Э.А., а группа ОН-- Э.Д. За счет того, что гидроксильная группа входит в общее р-π сопряжение с бензольным кольцом, в молекуле фенола происходит делокализация электронной плотности и кислотность увеличивается, т.к. сопряжение всегда сопровождается усилением кислотных свойств. Увеличение углеводородного радикала в монокарбоновых кислотах также влияет на изменение кислотных свойств и при введении заместителей в углеводородный происходит изменение кислотных свойств. Пример: в карбоновых кислотах при диссоциации образуются карбоксилат-ионы – самые стабильные органические анионы.
В карбоксилат-ионе отрицательный заряд за счет р, π-сопряжения распределён поровну между двумя атомами кислорода, т.е. он делокализован и соответственно менее концентрирован, поэтому в карбоновых кислотах кислотный центр более сильный, чем в спиртах и фенолах.
С увеличением углеводородного радикала, который выполняет роль Э.Д. кислотность монокарбоновых кислот снижается за счет уменьшения δ+на атоме углерода карбоксильной группы. Поэтому в гомологическом ряду кислот самой сильной является муравьиная кислота.
При введении Э.А. заместителя в углеводородный радикал, например хлора - кислотность соединения увеличивается, т.к. за счет –Iэффекта происходит делокализация электронной плотности и δ+на атоме С карбоксильной группы увеличивается, поэтому в данном примере трихлоруксусная кислота будет самой сильной. 3. Влияние растворителя. Взаимодействие молекул или ионов растворенного вещества с растворителем называется процессом сольватации. Стабильность аниона существенно зависит от его сольватации в растворе: чем больше ион сольватирован, тем он устойчивее, а сольватация тем больше, чем меньше размер иона и чем меньше делокализация в нем отрицательного заряда. . 3. Основные свойства органических соединений. π-основания и n-основания. Примеры нуклеофильных реакций Нуклеофильное замещение:
Механизм нуклеофильного замещения обозначается символом SN (по первым буквам английских терминов:S – substitution [замещение], N – nucleophile [нуклеофил]).
Нуклеофильное присоединение:
Обозначение механизма - AdN (Ad – addition [присоединение]). Опыт 1. Открытие уксусной кислоты Уксусная кислота оказывает бактерицидное и бактериостатическое действие. Например, 3 %-ный раствор уксусной кислоты убивает палочки брюшного тифа, 4 %-ный раствор – кишечную палочку. Особенно активна уксусная кислота по отношению к стафилококкам, служащим причиной пищевых отравлений. В пробирку поместите по 3 капли уксусной кислоты и воды. Испытайте реакцию раствора на лакмус. К раствору прибавьте 2-3 капли 10 %-ного раствора гидроксида натрия до полной нейтрализации уксусной кислоты. После этого добавьте 2-3 капли 1 %-ного раствора хлорида железа (III). Появляется желто-красное окрашивание за счет образования ацетата железа (III). Подогрейте раствор до кипения. Выделяется красно-бурый осадок не растворимого в воде гидроксида диацетата железа. Раствор над осадком становится бесцветным. Вопросы 1. Напишите схему реакции диссоциации уксусной кислоты. Как подтвердить этот процесс экспериментально? 2. Напишите схему реакции взаимодействия уксусной кислоты с гидроксидом натрия. Как экспериментально определить момент нейтрализации уксусной кислоты? 3. Напишите схему реакции образования ацетата железа (III). 4. Напишите структурную формулу гидроксида диацетата железа (III). Опыт 3 Образование нерастворимых солей высших жирных кислот. Ход работы: В пробирку последовательно поместите 5-6 капель раствора мыла и 1 - 2 капли раствора хлорида кальция. Хорошо перемешайте содержимое пробирки до образования белого осадка. В выводе отметьте: 1. Напишите уравнение реакции образования кальциевой соли стеариновой кислоты. 2. Какие соединения называют мылами?. 3. В состав каких биологически важных соединений входят высшие жирные кислоты? Опыт 4 Открытие этилового спирта реакцией образования уксусно-этилового эфира. Ход работы: В пробирку поместите половину лопаточки порошка безводного ацетата натрия и 4 капли этанола. Добавьте 3 капли концентрированной серной кислоты и осторожно нагрейте смесь до кипения. Через несколько секунд появляется характерный приятный запах этилацетата. Вопросы 1. Напишите уравнение реакции образования этилацетата с указанием механизма. 2.Какова роль концентрированной серной кислоты в реакции этерификации? Ход работы 1. Изолируем эритроциты крысы, проливая холодным (4 0С) изотоническим раствором хлористого натрия (0,15 М NaCl). Эритроциты суспензируем в0,15 М NaCl из расчета 1 мл эритроцитов / 6 объемов раствора NaCl, осаждаем эритроциты центрифугированием: 1000 оборотов/мин, 5 минут. Процедуру повторяем 3 раза. 2.Готовим суспензию эритроцитов с 10 % гематокритом. 1) контрольный образец: 2 мл, 2) опытный образец: 2 мл, 3) проба сравнения: 2 мл0.15 Мраствора NaCl. 3. Подвергаем опытный образец окислительному стрессу, используя органический трет–бутилгидропероксид: ТВООН к 2 мл суспензии эритроцитов (образец 2) прибавляем 20 мкл раствора ТВООН (100 мМ), создавая концентрацию окислителя 1 мМ. Инкубируем образец при 22 0С. Органический гидропероксид, взаимодействуя с ионами железа оксигемоглобина эритроцитов, образует алкоксильный ТВО. и пероксильный ТВОО. радикалам. Восстановленный глутатион в эритроцитах эффективно расходуется в реакциях детоксикации свободных радикалов, превращаясь в свою окисленную форму. Это происходит в реакции, катализируемой глутатион–пероксидазой (семейство селен–зависимых ферментов). 4. Для освобождения низкомолекулярных внутриклеточных компонентов разрушим клеточные мембраны и денатурируем белки клетки трихлоруксусной кислотой. К образцам (1), (2) и (3) добавим 0.2 мл 20 % трихлоруксусной кислоты. Для осаждения денатурированных белков и обрывков мембран подвергнем образцы центрифугированию. Надосадочная жидкость прдставляет раствор кислотно–растворимых низкомолекулярных компонентов клетки. 5. Определим в надосадочной жидкости содержание восстановленного глутатиона GSH. Предварительно приведем значение рН среды к нейтральному. Для этого к 1 мл супернатанта (надосадочной жидкости) добавим 1 мл 0,5 Мнатрий–фосфатного буфера, рН 7,8. Среди различных реагентов, используемых для специфического определения сульфгидрильных групп, наибольшую популярность приобрел реагент Эллмана: 5,5 – дитиобис (2–нитробензойная кислота)(Ellman, 1959), дисульфидное соединение. При рН 7.8–8.0 в реакциях с SH–группами реагент Эллмана образует окрашенный анион нитротиофенолята и смешанный дисульфид:
К 2 мл раствора добавим 30 мкл реактива Эллмана, концентрации 6. Определим оптическую плотность образцов (1) и (2), используя образец (3) в качестве кюветы сравнения (это так называемая проба на реактивы, содержащая все реагенты за исключением определяемого глутатиона). 7. Рассчитаем концентрацию внутриклеточного восстановленного глутатиона с учетом гематокрита суспензии и внесенных реагентов (разбавление образца): [GSH]= Д412 / 13600 ∙10 ∙ 1,1 ∙2 ∙1,03 Опишите полученный результат и принципы метода. Ход работы Навески НАДФ и изоцитрата растворяют перед приготовлением инкубационной среды. В пробу вносят компоненты инкубационной среды, которые заранее смешивают в указанных пропорциях в объеме, зависящем от количества проб.
В кювету вносят по 2,9 мл готовой инкубационной среды, затем, не вынимая кювету из прибора, добавляют 0,1 мл суспензии митохондрий или цитоплазмы, перемешивают. Реакцию начинают внесением препарата, содержащего фермен (0,1 млцитоплазмы либо суспензии митохондрий). Изменение оптической плотности раствора в результате восстановления НАДФ регистрируют в течение 3 - 5 мин с интервалом в 1 мин Активность фермента рассчитывают по формуле:
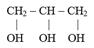
где ∆ Е – изменение оптической плотности за 1 мин; 3 – объем исследуемой пробы; 1000 – коэффициент перевода микромолей в наномоли; б – содержание белка в исследуемом образце, мг; 6,22 – коэффициент микромолярной экстинкции НАДФН (мМ-1 см-1). Общая характеристика. Полифункциональными называют соединения, в молекулах которых имеется несколько одинаковых функциональных групп. Среди полифункциональных соединений, участвующих в процессах жизнедеятельности, наиболее широко представлены соединения с гидроксильными и карбоксильными функциональными группами. Специальный интерес представляют β-дикарбонильные соединения. Соединения с несколькими аминогруппами встречаются реже. Многоатомные спирты и фенолы. Двухатомные спирты, т. е. спирты, содержащие две гидроксильные группы, имеют общее название диолы, или гликоли; трехатомные спирты называют триолами. Представителями таких спиртов являются этиленгликоль и глицерин соответственно. Общее название многоатомных спиртов - полиолы. В состав многих природных соединений входят в виде фрагментов двухатомные фенолы - пирокатехин, резорцин, гидрохинон.
этиленгликоль глицерин пирокатехин резорцин гидрохинон Этиленгликоль (этандиол-1,2) - высокотоксичная жидкость Глицерин (пропантриол-1,2,3) - нетоксичная вязкая жидкость сладкого вкуса (т. пл. 17 оС, т. кип. 290 оС), входит в состав большинства липидов. Применяется как компонент мазей для смягчения кожи. Пирокатехин (о-дигидроксибензол), называемый также катехолом, является структурным фрагментом многих биологически активних веществ, в частности катехоламинов. Монометиловый эфир пирокатехина – гваякол - применяется как компонент в составе лекарственных средств при катаре верхних дыхательных путей. Резорцин (м-дигидроксибензол) используется как антисептик и дезинфицирующее средство при кожных заболеваниях. Гидрохинон (п-дигидроксибензол), обладающий восстановительной способностью, является структурным фрагментом ряда соединений. В организме восстановительная способность замещенного гидрохинонового фрагмента делает его участником важного процесса транспорта электронов от окисляемого субстрата к кислороду. К спиртам высшей атомности относятся пентиты и гекситы, т. е. соответственно пяти- и шестиатомные спирты с открытой цепью. Накопление гидроксильных групп в молекуле ведет к появлению сладкого вкуса. Представители пентитов и гекситов – ксилит и сорбит - заменители сахара для больных диабетом. Многоатомный циклический спирт миоинозит относится к витаминоподобным соединениям (витамины группы В) и является структурным компонентом сложных липидов - фосфатидилинозитов. В растениях широко распространена фитиновая кислота, представляющая собой гексафосфат миоинозита. Кальциевая или смешанная кальций-магниевая соль фитиновой кислоты, называемая фитином, улучшает состояние нервной системы при заболеваниях, связанных с недостатком фосфора в организме. Дикарбоновые кислоты. Карбоновые кислоты, содержащие в своем составе одну карбоксильную группу, называют одноосновными, две - двухосновными и т. д. В настоящем разделе будут рассмотрены некоторые представители дикарбоновых кислот алифатического и ароматического рядов (таблица). Все они представляют собой кристаллические вещества. Таблица. Названия некоторых дикарбоновых кислот и их производных
Систематические названия дикарбоновых кислот строятся по общим правилам заместительной номенклатуры. Однако для боль- шинства из них предпочтительны тривиальные названия. Их латинские названия служат основой названия анионов и производных кислот, которые часто не совпадают с русскими тривиальными названиями (см. табл.). Щавелевая кислота - простейшая двухосновная кислота. Некоторые ее соли, например оксалат кальция, трудно растворимы и часто образуют камни в почках и мочевом пузыре (оксалатные камни). Янтарная кислота в заметном количестве была обнаружена в янтаре, откуда получила название сама кислота и ее производные сукцинаты (от лат. succinium - янтарь). Малеиновая и фумаровая кислоты - представители ненасыщенных дикарбоновых кислот с одной двойной связью. Фумаровая кислота участвует в обменных процессах, протекающих в организме.
Диамины. Наиболее известны тетраметилендиамин, или путрес- цин H2N(CH2)4NH2, и пентаметилендиамин, или кадаверин H2N(CH2)5NH2. Их долгое время считали трупными ядами, т. е. веществами, образующимися при декарбоксилировании диаминокислот и обусловливающими ядовитость гниющих белков. В настоящее время выяснено, что ядовитые свойства белкам при гниении придают другие вещества. Общая характеристика Большинство веществ, участвующих в метаболизме, являются гетерофункциональными соединениями. Гетерофункциональными называют соединения, в молекулах которых имеются различные функциональные группы. Характерные для биологически важных соединений сочетания функциональных групп представлены в таблица 3.2. Таблица 3.1. Наиболее распространенные сочетания функциональных групп в биологически важных алифатических соединениях
Среди гетерофункциональных соединений в природных объектах наиболее распространены аминоспирты, аминокислоты, гидроксикарбонильные соединения, а также гидрокси- и оксокислоты (табл. 9.2). Таблица 9.2. Некоторые гидрокси- и оксокислоты и их производные
* Для ди- и трикарбоновых кислот - при участии всех карбоксильных групп. Для неполных солей и функциональных производных добавляется префикс гидр(о)-, например «гидроксалат» для аниона НООС-СОО-. Имеющие особую биологическую важность α-аминокислоты описаны в главе 12. Полигидроксиальдегиды и полигидроксикетоны (углеводы) рассматриваются в главе 13. В ароматическом ряду основу важных природных биологически активных соединений и синтетических лекарственных средств (см. 9.3) составляют и-аминофенол, и-аминобензойная, салициловая и сульфаниловая кислоты.
Систематические названия гетерофункциональных соединений строятся по общим правилам заместительной номенклатуры (см. 1.2.1). Однако для ряда широко распространенных кислот пред
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-16; просмотров: 786; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.15 (0.014 с.) |






















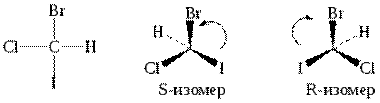






 увеличение Э.О. и кислотности
увеличение Э.О. и кислотности увеличение кислотности
увеличение кислотности