
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Нарушения метаболизма у людей, страдающих ожирением.Содержание книги
Похожие статьи вашей тематики
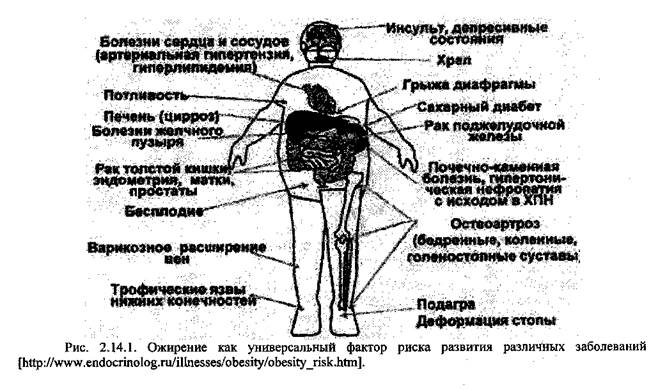
Поиск на нашем сайте У людей с избыточным количеством жировой ткани имеются следующие общие отклонения от нормального обмена веществ: • снижена толерантность к глюкозе (может восстанавливаться при снижении массы тела); • гипертриглицеридемия в результате гиперлшюпротеинемий II-V типов, чаще всего повышена концентрация ЛПОНП и ЛПНП; • гиперинсулинемия; • снижена продукции гормона роста. На рисунке 2.14.1. схематически представлена взаимосвязь ожирения с развитием полиорганной патологии.
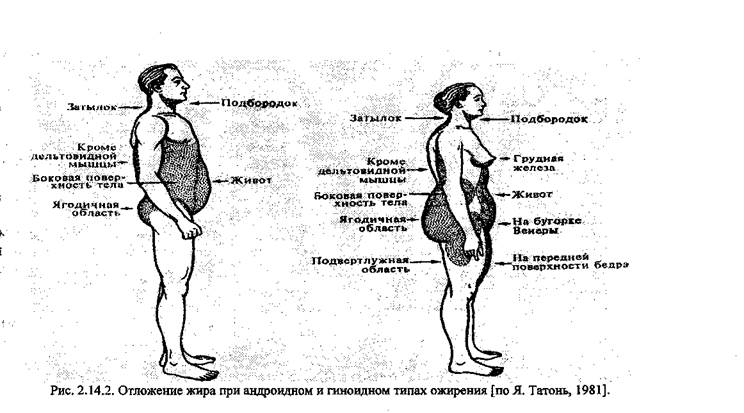
Классификация ожирения. I. По этиологии 1. Первичное (алиментарно-конституциональное, экзогенно-конституциональное). 2. Вторичное (симптоматическое). а) Церебральное ожирение • опухоли головного мозга • травмы мозга и последствия хирургических операций • синдром пустого турецкого седла • воспалительные заболевания мозга (энцефалит и др.) Ь) Эндокринное ожирение • гипофизарное • гипотиреоидное • климактерическое • надпочечниковое с) Ожирение на фоне психических заболеваний и/или приема нейролептиков. II. По типу отложения жира (рис.2.14.2;) 1. Генерализованное и местное. 2. Андроидное (по мужскому типу). 3. Гиноидное (по женскому типу). III. По характеру жировой ткани 1. Гипертрофическое (увеличение размера жировых клеток). 2. Гиперпластическое (увеличение количества жировых клеток). 3. Смешанное. IV. По степени тяжести Степень ожирения рассчитывается как отношение должной массы тела к измеренной и умноженное на 100%: I. 15-29% И. 30 - 49% III. 50-99% IV. 100% и выше
Первичное ожирение (или алиментарно-конституциональное) развивается при избытке поступающей в организм с пищей энергии по сравнению с ее расходом. Патогенез первичного ожирения. 1. Алиментарный дисбаланс (избыточная энергетическая ценность питания) - веду Контроль за отложением энергетических субстратов (триацилглицеринов) в ади-поцитах и их мобилизацией происходит с участием гормонов. После еды при увеличении концентрации глюкозы в крови синтез жиров под влиянием инсулина в адипоцитах увеличивается. При первичном ожирении возможен функциональный гиперинсулинизм. 2. Нарушение регуляции чувства голода и насыщения Количество потребляемой пищи зависит от регуляции чувства голода и насыщения, которые определяются концентрацией в крови глюкозы и гормонов пищеварительного тракта, которые инициируют чувство насыщения (холецистокинин-панкреозимин, ней-ротензин, бомбезин). Выделяясь в кровь при поступлении пищи в пищеварительный тракт, они возбуждают центр насыщения (вентро-медиальные ядра гипоталамуса) и тормозят центр голода (латеральные ядра гипоталамуса). Аналогичные эффект вызывает растяжение желудка пищей и возбуждение его рецепторов вазоактивным интерстициальным полипептидом, холецистокинином-панкреозимином. При первичном ожирении нарушается выработка данных веществ, а желудок растянут (имеет большой объем), поэтому требуется значительный объем пищи для возбуждения его рецепторов растяжения. Может страдать периферический механизм (снижение чувствительности рецепторов желудка и гипоталамуса к гормонам пищеварительного тракта). 3. Наследственность Ген ожирения (obese gene) обнаружен у человека и других млекопитающих. Одиночные мутации в этом гене приводят к избыточной массе тела. Продуктом экспрессии гена оЬ является белок оЬ (или «лептин», от греч. leptos - тонкий), он секретируется в кровь адипоцитами. Одним из органов-мишеней белка оЬ является центральная нервная система, через которую гормон осуществляет свое действие. Белок оЬ увеличивает энергетический обмен, потребление кислорода, температуру тела, двигательную активность, снижает потребление пищи —> уменьшается масса тела вследствие снижения количества жиров в организме. 4. Нарушение чувствительности к липолитическим гормонам: адреналин, глюкагон, СТГ, тироксин. 5. Культурный фактор - национальные и семейные традиции питания (предпочтительное употребление богатой жирами пищи в северных районах, привычки «заедать» просмотр кинофильма, чтение книг, работу на компьютере и т.д.) Теории регуляции потребления пищи:
1. Глюкостатическая теория - повышенное потребление пищи обусловлено снижением чувствительности глюкорецепторов центра насыщения. 2. Липостатическая теория - повьппенное потребление пищи обусловлено повышением в крови концентрации жирных кислот и стимуляции ими центра голода. Концентрация ЖК в плазме повышается при голодании (мобилизация жирных кислот из адипоцитов), а также при приеме пищи, богатой жирами. Вторичное ожирение (или симптоматическое ожирение), 1. Гипофизарное ожирение (болезнь Иценко-Кушинга, центральный гиперкортицизм) Характерно лунообразное багрово-красное лицо, гипертрихоз, стрии, увеличение АД, гипергликемия. Ожирение носит диспластических характер. Жироотложения выявляются преимущественно в области верхней половины туловища и лице, при худых конечностях. Глюкокортикоиды вызывают гипергликемию —» избыток глюкозы утилизируется преимущественно инсулинзависимыми тканями (в частности, жировой и особенно зонами с наибольшим количеством рецепторов к инсулину - верхняя половина туловища, лицо) -» избыток поступившей в адипоциты глюкозы превращается в триглицериды и откладывается. 2. Надпочечниковое ожирение (синдром Иценко-Кушинга, первично-гландулярная форма гиперкортицизма - опухоль пучковой зоны коры надпочечников) развивается при повышенной продукции глюкокортикоидовю. Продукция АКТГ при этом заболевании снижается по принципу обратной связи. Механизм ожирения аналогичен таковому при болезни Иценко-Кушинга. Наблюдается одностороннее увеличение надпочечника при гипотрофии другого. 3. Гипотиреоидное ожирение является следствием недостаточности гормонопродуци-рующей функции щитовидной железы. Дефицит тироксина и трийодтиронина приводит к снижению интенсивности основного обмена. 4. Ожирение при опухоли b-клеток островкового аппарата поджелудочной железы (секретирующая инсулинома) развивается вследствие избытка инсулина, который тормозит липолиз и стимулирует липогенез. Характерна гипогликемия. 5. Гипоовариальное и климактерическое ожирение является результатом снижения секреции половых гормонов —> снижение основного обмена преобладание анаболических процессов. Снижение секреции гонадотропинов, обладающих липотропным действием, —► снижение процессов липолиза. 6. Ожирение при адипозогенитальной дистрофии связанно с поражением гипоталамо-гипофизарной системы (внутриутробная инфекция - токсоплазмоз; перинатальная травма; опухоли - краниофарингиома, хромофобная аденома; тромбозы; эмболии; кровоизлияния) и характеризующееся недоразвитием половых желез и ожирением. При поражении гипоталамуса происходит повреждение или раздражение его пара-вентрикулярных и вентромедиальных ядер, что ведет к резкому повышению аппетита с последующим развитием ожирения. Вследствие поражения гипоталамуса снижается гона-
IV. Нарушение платочного метаболизма липидов. Жировая инфильтрация органов. Жировая инфильтрация - избыточное отложение жиров в тканях, не относящихся к жировой. На рисунке 2.1 4.3. схематически представлен патогенез жировой инфильтрации печени.
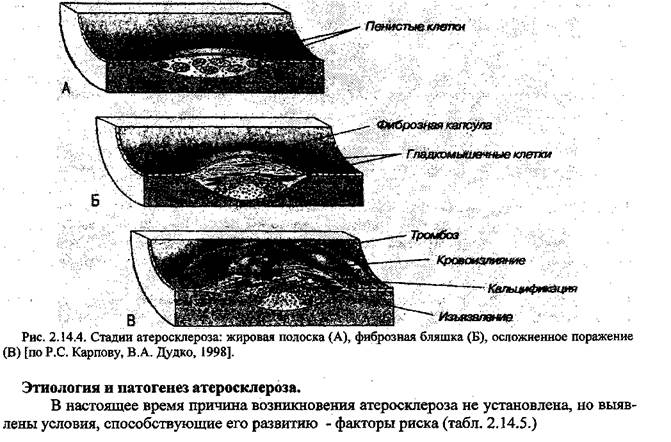
Рис. 2.14.3. Схема патогенеза жировой инфильтрации печени. V Этиология и патогенез атеросклероза. Стадии развития атеросклероза и осложнения. Атеросклероз («athere» - кашицеобразная масса (греч.), «sclerosis».- твердый) -хроническое заболевание, характеризующееся поражением стенки артериальных сосудов, в основе которого лежит нарушение липидного обмена, отложение в сосудистой стенке жировых масс с утолщением и деформацией стенки. Атеромы вызывают стенозы (сужение просвета сосуда) и окклюзии (закупорку сосуда) артерий, что приводит к нарушениям гемодинамики в дистальном артериальном русле, ишемии и гипоксии органов и тканей. При осложненных атеромах развиваются тромбозы и эмболии. Осложнения атеросклероза являются основной причиной смерти: ишемическая болезнь сердца (стенокардия) и инфаркт миокарда (60% мужчин и 41% женщин), ишемическая болезнь мозга с нарушением мозгового кровообращения, ишемическим и геморрагическим (кровоизлияние в мозг) инсультом (25% мужчин и 39% женщин, 1991). Частым проявлением атеросклероза является ишемия нижних конечностей, проявляющаяся симптомом «перемежающей хромоты» (ограничение дистанции ходьбы из-за болей в конечности с восстановлением ходьбы и снижением болевого синдрома после отдыха). Грозным осложнением атеросклероза является аневризма аорты (расширение аорты с деградацией ее стенки), которая может привести к разрыву стенки аорты с несовместимым с жизнью кровотечением. Атеросклеротическое поражение почечных артерий приводит к развитию стойкой вазоренальной артериальной гипертензии, а атеросклероз брыжеечных артерий приводит к ишемии кишечника (ишемический абдоминальный синдром) Атеросклероз поражает аорту и крупные артерии эластического типа, преимущественно в местах их бифуркацию. Стадии развития атеросклероза. 1. Долипидная стадия - создаются условия для проникновения липидов в интиму, обна Долипидная стадия объективно проявляется снижением эластичности артерий (увеличение скорости распространения пульсовой волны) и дисфункцией эндотелия (в основном снижение выработки эндотелием оксида азота). 2. Стадия липоидоза (жировые пятна и полоски) начинается с накопления в интиме Стадия липидных пятен и полосок может быть установлена по утолщению комплекса «интима - медиа» дистального сантиметра общей сонной артерии, измеренного при помощи ультразвукового исследования. 3. Стадия липосклброза проявляется разрастанием молодой соединительной ткани в участках отлоения липопротеидов, созревание соединительной ткани ведет к образованию фиброзных бляшек. Вследствие нарушений эндотелия на поверхности интимы в местах скопления липидов может произойти агрегация тромбоцитов или выпадение нитей фибрина, что, является началом фибропластического процесса. Дальнейшее прогрессирующее накопление липидов сопровождается выраженым измением метаболизма гладко-мышечных клеток сосудов. Они теряют способность утилизировать поступающие жиры, которые по мере гибели клеток попадают в окружающие ткани, где располагаются в основном веществе, вдоль эластических волокон. Высвободившиеся липиды и фибриноген пропитывают бляшку, последний оказывает фибропластическое и склерозирую-щее действие. Возникает типичная фиброзная бляшка, состоящая из бедной клеточными элементами грубой соединительной ткани и жировых масс. 4. Стадия атероматоза (осложненная бляшка) характеризуется распадом в зоне бляшки липоидов, коллагеновых волокон, а также мышечных и ксантомных клеток. В результате образуется полость, содержащая жиро-белковый детрит (атероматозные массы) и от- деленная от просвета сосуда прослойкой соединительной ткани, которая играет роль покрышки бляшки. Прогрессирование атероматоза приводит к осложненным поражениям сосудов -кровоизлияниям в бляшку, разрушению ее покрышки и изъязвлению. Выпадающий при этом в просвет сосудов детрит может стать источником эмболии, а сама атероматозная язва - служить основой для образования тромбов. В процессе разрушения клеток и кровоизлияния в атерому, в ней может накапливаться кальций, что обозначается как кальцинирование бляшки и имеет характерный вид. 5. Стадия атёрокальцйноза - отложение солей кальция в атероматозные массы, межуточное вещество и фиброзную ткань (петрификация). На рисунке 2.14.4. схематически представлена последовательность морфологических изменений в сосудистой стенке при атеросклерозе.
Существует большое многообразие теорий развития атеросклероза. Многие из них имеют историческую ценность. В настоящее время общепризнано, что основными звеньями патогенеза атеросклероза являются: нарушения метаболизма липидов, повреждение эндотелия и воспаление. Первой научной теорией развития атеросклероза является инфильтрационно-комбинационная (холестериновая) теория атеросклероза Н.Н. Аничкова [1915,1935]. Она базируется на положении, согласно которому основная часть энергетических потребностей артериальной стенки, особенно ее бессосудистых структур (интимы и внутренней трети медии), восполняется за счет липидов плазмы крови. Делается допущение, что плазменные липиды поступают в сосудистую стенку путем просачивания (инфильтрации) плазмы в направлении от эндотелия к адвентиции. Предполагается также, что в норме липиды просачивающейся плазмы проходят без задержки в адвентицию и удаляются через систему лимфатических сосудов. Однако, когда количество липидов велико, они накапливаются в сосудистой стенке, вызывая развитие липидоза. Весомым подтверждением холестериновой концепции заболевания являются случаи гомозиготной гиперхолестеринемии, при которой тяжелейший атеросклероз развивается в юношеские и даже детские годы, и только снижение уровня холестерина в крови, каким бы путем оно ни достигалось, спасает больных от инфаркта миокарда и неминуемой гибели. В настоящее время эта теория получила дальнейшее развитие. Прежде всего, установлено, что развитию атеросклероза способствует нарушение метаболизма в организме липопротеидов с преобладанием в крови атерогенных липопротеидов - ЛПНП и ЛПОНП, снижение концентрации ЛПВП. Дело в том, что физиологическая функция ЛПНП заключается в транспорте холестерина в клетки, а ЛПВП наоборот выводит избытки холестерина из клеток На этом основании для оценки атерогенности плазмы крови был предложен коэффициент атерогенности Климова (КА), который в норме должен быть ниже 3,5 у.е., а его увеличение отражает возрастание риска развития атеросклеротическо-го поражения артерий. В приведенной ниже формуле для расчета КА, ОХС - концентрация в плазме крови общего ХС, ХС ЛПВП - холестерина ЛПВП. ОХС-ХСЛПВП хслпвп [ ____:________ I Установлено так же, что белковая часть молекулы липопротеидов (апопротеины) играет важную роль во взаимодействии этих молекул с клетками, обеспечивая связь молекулы с соответствующим рецептором клетки для ЛПНП, или ЛПВП. Поэтому избыток апопротеина В (апо В), входящего в состав ЛПНП, способствует избыточному транспорту ХС в клетки, а недостаток апо А, входящего в состав ЛПВП, препятствует его выведению из клеток и из организма в целом. Существенный вклад понимание природы атеросклеротического процесса внесла аутоиммунная теория А.Н. Климова и др. (1980-1995), Согласно этой теории инициацию атеросклероза вызывают не столько липопротеины, сколько аутоиммунные комплексы, содержащие липопротеины в качестве антигена. Аутоиммунные комплексы вызывают повреждение эндотелия, ускоряют проникновение липопротеидов в сосудистую стенку; продлевают циркуляцию липопротеинов в крови и задерживают окисление и экскрецию холестерина с желчью, способствуют развитию гиперлипопротеинемии; откладываясь и фиксируясь в стенке артерий, оказывают цитотоксическое действие. В настоящее время популярной является воспалительная теория атеросклероза. В ее основе лежит сходство атеросклеротического процесса с воспалением. При атеросклерозе отмечена выраженная лейкоцитарная инфильтрация сосудистой стенки; активация макрофагов, фагоцитоз липопротеидов, холестерина, аутоиммунных комплексов, обломков клеточных структур; выделение лейкоцитами провосполительных цитокинов; гипергидратация сосудистой стенки и пролиферация гладкомышечных клеток. Воспалительная теория атеросклероза позволяет понять, что агрессивное развитие атеросклероза с формированием осложненных тромбо- и эмболоопасных бляшек связано с преобладанием провоспалительных цитокинов и гиперэргическим течением воспаления в сосудистой стенке, а стабильное течение атеросклероза - с преобладанием противовоспалительных цитокинов и гипоэргическим воспалением. Важную роль в патогенезе атеросклероза играет повреждение эндотелия артериальной стенки. Эндотелий играет роль защитного барьера в отношении атеросклероза. Повреждение эндотелия вследствие воздействия гемодинамических факторов (кровяное давление, турбулентность потока крови, ее боковое давление), токсинов (никотин, некоторые лекарственные препараты), воспалительных процессов (вирусных, бактериальных, иммунологических), образования тромбов способствует проникновению макромоле-кулярных соединений из плазмы крови в артериальную стенку. Повреждение эндотелия сопровождается развитием дисфункции эндотелия, которая проявляется нарушением выработки в эндотелии биологически активных соединений. Важное значение при этом имеет снижение выработки эндотелиальными клетками оксида азота, оказывающего сосудорасширяющее действие, и цростациклина, снижающего тромборезистентность сосудистой стенки. Кроме того, в процессе развития атеросклероза значимую роль играет акцивация пе-рекисного окисления линидов. Свободно-радикальное окисление ненасыщенных жирных кислот вызывает повреждение клеточных мембран в сосудистой стенке и способствует развитию атеросклероза. Перекиси липидов ингибируют в эндотелиальных клетках артерий фермент про-стациклин-синтетазу в результате развивается локальная недостаточность простациклина при сравнительно высоком содержании тромбоксана, что усиливает агрегацию тромбоцитов на поверхности эндотелия, способствует развитию атеросклероза и его тромботиче-ских осложнений. В целом нарушение свертывающей системы крови с гиперкоагуляцией способствует развитию атеросклероза и атеротромбоза. Существенное стимулирующее влияние на развитие атеросклероза оказывает нарушение метаболизма аминокислоты гомоцистеина - гипергомоцистеинемия. Гомоци-стеин - высокореакционная аминокислота, которая легко подвергается аутоокислению, что запускает в крови каскад свободно-радикального окисления, вызывающий повреждение эндотелия. Токсическое повреждение эндотелия сопровождается увеличением пролиферации гладкомышечных клеток, усилением адгезии и агрегации тромбоцитов, активацией коагуляционного гемостаза и торможением фибринолиза, дислипоппротеидемией, усилением адгезии моноцитов к эндотелию, увеличением продукции провоспалительных цитокинов (ФИО, ИЛ 6, 12). Все это приводит к ускоренному развитию атеросклероза с агрессивным течением. При гипергомоцистеинемии атеросклероз дебютирует на 5-10 лет раньше обычного срока (у людей 30-40 лет) и протекает с высокой склонностью к тромбо-тическим осложнениям. Гомоцистеин образуется в организме из метионина, который поступает с пищей. Избыток гомоцистеина превращается в цистеин и выводится с мочой, и (или) вновь превращается в метионин. Превращения метионина протекают под влиянием ферментов, кофакторами которых служат витамины Вб, В12 и фолиевая кислота. Поэтому основной причиной гипергомоцистеинемии является витаминодефицит при нарушении питания, пищеварения и алкоголизме. Значительную роль играет аутосомно-рецессивная наследственная ферментопатия энзимов метаболизма гомоцистеина. К гипергомоцистеинемии приводит так же почечная недостаточность. Патология белкового обмена. Белок - высокомолекулярный органический биополимер, состоящий в основном из аминокислот, соединенных пептидной связью. Классификация белков. I. По растворимости: ■ водорастворимые, ■ солерастворимые, ■ спирторастворимые, ■ нерастворимые, ■ прочие. П. По конформационной структуре: ■ фибриллярные (коллагены, эластины, кератины), ■ глобулярные: альбумины, глобулины, гистоны (все ферменты и большинство БАВ). III. По химическому строению: 1. Простые (протеины) - состоят только из аминокислот. 1.1. Глобулярные а) Альбумины - растворимы в воде, не растворимы в концентрированных растворах солей. b) Глобулины - не растворимы в воде, растворимы в солевых растворах, с) Гистоны - растворимы в воде, в слабоконцентрированных кислотах. Обладают выраженными основными свойствами. Это ядерные белки, они связаны с ДНК и РНК. d) Протамины - ассоциациированы с нуклеиновыми кислотами. 1.2. Фибриллярные (склеропротеины) ■- белки опорных тканей (хрящей, костей), шер ■ коллагены - фибрилярные белки соединительной ткани. При длительном кипячении они растворяются в воде и при застудневании образуется желатин. ■ эластины — белки связок и сухожилий. По свойствам похожи на коллагены, но подвергаются гидролизу под действием ферментов пищеварительного сока; ■ кератин - входит в состав волос; 2. Сложные (протеиды) - помимо аминокислот имеют в составе небелковую часть а) Нуклеопротеиды ~ простетическая група - нуклеиновые кислоты. Среди многочисленных классов нуклеопротеидов наиболее изученными являются рибосомы, состоящие из нескольких молекул РНК и рибосомных белков, и хроматин - основной нуклеопротеид эукариотических клеток, состоящий из ДНК и структурообразующих белков - гистонов (содержатся в клеточном ядре и митохондриях). b) Гемопротеиды - небелковый компонент этих протеидов — тем, построен из четырех пиррольных колец, с ними связан ион двухвалентного железа (через атомы азота). К таким белка относятся: гемоглобин, миоглобин, цитохромы. Этот класс белков еще называют хромопротеиды, поскольку гем является окрашенным соединением. Гемоглобин - транспорт кислорода. Миоглобин - запасание кислорода в мышцах. Цитохромы (ферменты) — катализ окислительно-восстановаительных реакций и электронный транспорт в дыхательной цепи. с) Металлопротеиды - в состав простетической группы входят металлы. Цитохром а - содержит медь, сукцинатдегидрогеназа и др. ферменты содержат негеминовое железо (ферродоксин). d) Липопротеиды ~ содержат липиды, входят в состав клеточных мембран е) Фосфопротеиды - содержат остаток фосфорной кислоты f) Глюкопротеиды - содержат сахара Функции белков: структурная (пластическая), каталитическая, транспортная, ме-ханохимическая, регуляторная, защитная, опорная, энергетическая, рецепторная. Потребность в белках. Дети 0,88-0,77 г/кг массы тела. Подростки 0,72-0,64 г/кг массы тела. Взрослые 0,59 г/кг массы тела. Человек массой 70 кг ежедневно потребляет с пищей около 80-100 г белка. Кроме того, 10-20 г белка секретируется в виде ферментов и еще приблизительно 20 г белка дают клетки слизистой оболочки, слущивающиеся с поверхности пищеварительного тракта. Практически весь этот белок переваривается и всасывается. Переваривание белков На первой стадии переваривания пища подвергается механическому измельчению в полости рта, что увеличивает площадь поверхности для последующих стадий. Соляная кислота, секретируемая париетальными клетками желудка убивает бактерии и вызывает денатурацию белков, что увеличивает поверхность, на которую воздействуют пищеварительные ферменты. Пищеварительные ферменты выделяются в полость желудочно-кишечного тракта в неактивной форме (зимогены), поэтому они не повреждают слой эпителиальных клеток слизистой оболочки, выстилающей полость изнутри. Пепсиноген (предшественник пищеварительного фермента пепсина) секретируется зимогенпродуцирующими клетками желудка. Пепсин сохраняет стабильность только в кислой среде желудочного содержимого, где он расщепляет пептидные связи. Возникающие в результате большие пептидные фрагменты и отдельные аминокислоты стимулируют секрецию пищеварительных ферментов в тонкую кишку. Переваривание белков в тонкой кишке начинается с регулируемого выделения эн-терокиназы эпителиальными клетками двенадцатиперстной кишки и зависит от секреции ионов бикарбоната, которые нейтрализуют кислоту, поступающую с желудочным содержимым. Энтерокиназа отщепляет гексапептид от молекулы трипсиногена (одного из зи-могенов, секретируемых поджелудочной железой), превращая его в трипсин. Трипсин обладает аутокаталитической активностью и, кроме того, активирует другие панкреатические зимогены, отщепляя от них пептидные фрагменты. Активированные ферменты поджелудочной железы гидролизуют пептидные связи в различных участках полипептидных цепей. Трипсин, химотрипсин и эластаза относятся к эндопептидазам, расщепляющим связи внутри цепи. Две карбоксипептидазы отщепляют аминокислоты от Оконца молекул белка. Олигопептиды, образующиеся в результате действия панкреатических ферментов, подвергаются дальнейшему расщеплению с помощью аминопептидаз и дипептидаз, расположенных на поверхности эпителиальных клеток кишечника. Конечными продуктами переваривания белков в полости кишечника являются аминокислоты, дипептиды и три-пептиды, которые всасываются клетками эпителия. Дальнейший гидролиз пептидных связей происходит внутри клеток эпителия перед окончательным транспортом аминокислот в кровь воротной системы. В целом все ди- и три- пептиды распадаются на составляющие их аминокислоты внутри клеток эпителия. Исключением являются пептиды, содержащие пролин, гидрокси-пролин или необычные аминокислоты. Всасывание аминокислот происходит в тонком отделе кишечника. Это активный процесс и требует затраты энергии. Аминокислоты попадают в портальный кровоток —> в
В толстом отделе кишечника, не всосавшиеся по каким-либо причинам пептиды и аминокислоты, подвергаются процессам гниения. При этом образуются такие продукты как фенол, крезол, сероводород, метилмеркаптан, индол, скатол, кадаверин, путресцин. Эти вещества всасываются в кровь и поступают в печень, где подвергаются конъюгации с глюкуроновой кислотой и другим процессам обезвреживания. Затем они выводятся из организма с мочой.
|
||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-02-08; просмотров: 639; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.214 (0.013 с.) |

 дотропная функция гипофиза. Это в свою очередь приводит к гипогонадизму с последующим изменением высшей нервной деятельности и развитием ожирения.
дотропная функция гипофиза. Это в свою очередь приводит к гипогонадизму с последующим изменением высшей нервной деятельности и развитием ожирения.


 печень и в общий кровоток. Печень и почки поглощают аминокислоты интенсивно; мозг избирательно поглощает метионин, гистидин, глицин, аргинин, глутамин, тирозин.
печень и в общий кровоток. Печень и почки поглощают аминокислоты интенсивно; мозг избирательно поглощает метионин, гистидин, глицин, аргинин, глутамин, тирозин.


