
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Схемы исследования новых лекарственных средствСодержание книги
Поиск на нашем сайте
После завершения III фазы клинических испытаний документация вновь поступает в Фармакологический комитет (объем полного досье может составлять до 1 млн. страниц) и в течение 1-2 лет регистрируется в Государственном реестре лекарственных средств и изделий медицинского назначения. Только после этого фармакологический концерн имеет право начать промышленный выпуск лекарственного средства и его распространение через аптечную сеть.
IV фаза клинических испытаний (постмаркетинговые исследования). Цель этой фазы – выявить редко встречающиеся, но потенциально опасные нежелательные эффекты лекарства. При организации и проведении клинических испытаний должны выполняться следующие требования: Исследование должно быть рандомизированным, контролируемым – т.е. параллельно с группой принимающей исследуемое лекарство, должна быть набрана группа, которая получает стандартный препарат сравнения (позитивный контроль) или неактивный препарат, который внешне имитирует изучаемое лекарство (плацебо контроль). Здесь используется метод "слепого контроля", эффект плацебо, метод двойного "слепого контроля", когда ни врач, ни больной не знает, когда это плацебо используется. Знает только специальная комиссия. Клинические испытания проводятся на людях, и во многих странах это осуществляется на добровольцах. Здесь, безусловно, возникает масса юридических, деонтологических, нравственных аспектов проблемы, которые требуют своей четкой разработки, регламентации и утверждения законов на данный счет. Международные стандарты GLP, GCP, GMP С целью повышения качества медицинской помощи была разработана серия надлежащих практик GxP. Система GxP охватывает все этапы жизненного цикла лекарственного средства, от фармацевтической разработки до конечного потребителя, а именно: · Доклинические (лабораторные) исследования, которые регулируются правилами GLP (Good Laboratory Practice, Надлежащая лабораторная практика), · Клинические испытания, которые регулируются правилами GCP (Good Clinical Practice, Надлежащая клиническая практика), · Производство, которое регулируется правилами GMP (Good Manufacturing Practice, Надлежащая производственная практика), GCP – надлежащая клиническая практика (Good Clinical Practice). Стандарт для клинических испытаний, охватывающий планирование, проведение, завершение, проверку, анализ результатов, составление отчетов и ведение документации, обеспечивающий научную значимость исследований, этическую приемлемость и полную документированность клинических характеристик терапевтического (диагностического, профилактического) продукта. GLP – надлежащая лабораторная практика (Good Laboratory Practice). Набор критериев, соблюдение которых необходимо в качестве основы оценки результатов и выводов лабораторных исследований.
· Хранение, которое регулируется правилами GSP (Good Service Practice, Надлежащая практика обслуживания, хранения), · Оптовая торговля, которая регулируется правилами GDP (Good Distribution Practice, Надлежащая практика оптовой продажи), · Розничная торговля, которая регулируется правилами GPP (Good Participatory Practice, Надлежащая практика розничной продажи).
|
||||||||||
|
|
Последнее изменение этой страницы: 2017-01-19; просмотров: 442; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.15 (0.008 с.) |

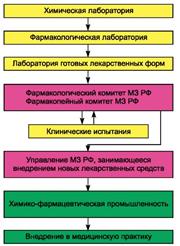 Краткая характеристика основных этапов при разработке новых лекарств.
Краткая характеристика основных этапов при разработке новых лекарств.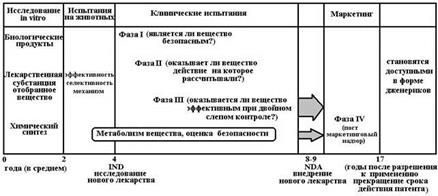
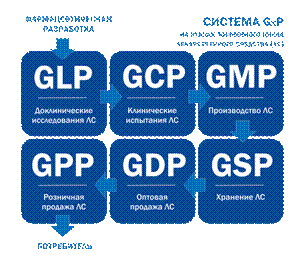 GMP – надлежащая производственная практика (Good Manifacturing Practice). Часть системы обеспечения качества, которая гарантирует, что продукция постоянно производится и контролируется по стандартам качества, соответствующим её назначению и требуемым торговой лицензией.
GMP – надлежащая производственная практика (Good Manifacturing Practice). Часть системы обеспечения качества, которая гарантирует, что продукция постоянно производится и контролируется по стандартам качества, соответствующим её назначению и требуемым торговой лицензией.


