
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Некоторые продукты многотоннажных производствСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Неорганические продукты: HCl (36 %); H2SO4 (92 – 96; 98, 100 %); SO3 (6 – 60 %); H3PO4; POCl3; PCl3; PCl5; PBr3; P2S5; P2O5; NH3 (30 – 34 % водный раствор; газ в баллонах); H2N-NH2×H2O; H2N-NH-CH3; H2N-N(CH3)2; HNO3 (63; 98 – 100 %); NaNO3; KNO3; NH4NO3; NaNO2; NaCN; S; Na2S; NH4NCS; NaNCS; SCl2; SOCl2; SO2Cl2; Cl2; Br2; I2; Ca(OCl)2; NaMnO4; KMnO4; CrO3; Na2Cr2O7; H2; LiAlH4; NaBH4; NaHSO3; Na2SO3; Al(i -OC3H7)3; NaOH; KOH; NaCl; KCl; Na2SO4; порошки – Cu; Zn; Fe; Al и др.
Органические продукты:
Наиболее широко используемые ароматические соединения
Безусловно, приведена лишь небольшая часть промежуточных продуктов. При разработке промышленного метода синтеза необходимо выяснить производителей и цену за единицу продукции.
1.4. ТЕОРЕТИЧЕСКИЕ АСПЕКТЫ ВЫБОРА МЕТОДА СИНТЕЗА
Классификация реагентов и реакций, механизмы реакций, их энергетика, энергетическая диаграмма реакции, уравнение Клопмана, принцип ЖМКО, кинетика, анализ общего кинетического уравнения, влияние заместителей и растворителей, уравнение Гаммета, квантово-химические расчеты.
При разработке оптимального плана синтеза субстанции лекарственного препарата используют весь богатый арсенал методов органической и физической химии, учитывают возможности их применения в технологически приемлемых условиях. Необходимо знать как сами методы, так и условия проведения реакций, закономерности их протекания, использующееся технологическое оборудование, способы выделения и очистки соединений. При рассмотрении закономерностей химических процессов необходимо знать классификацию реагентов и реакций, механизмы реакций, их энергетику, кинетику, влияние заместителей и растворителей.
1.4.1. Энергетические факторы
Хорошо известно, что реакции протекают в результате эффективного соударения двух частиц. Атакуемую частицу называют субстратом реакции, атакующую – реагентом. Возможность прохождения реакции с точки зрения термодинамики контролируется изменением свободной энергии системы DG = DH - TDS, где DH, DS – энтальпийная и энтропийная составляющие. Если свободная энергия исходной системы выше свободной энергии продуктов реакции, то принципиально реакция может идти без подвода энергии извне. Однако при этом скорость реакции может быть ничтожно мала. Для того чтобы осуществить реакцию с заметной скоростью, необходимо преодолеть энергетический барьер, называемый свободной энергией активации (DG ¹). Тогда возникает переходное состояние либо образуется промежуточное соединение, либо конечный продукт. В переходном состоянии наблюдается частичное образование связи, в промежуточном соединении и конечном продукте возникает новая ковалентная связь. Эту ситуацию можно отразить с помощью энергетической диаграммы реакции. Для некоторых реакций замещения в ряду ароматических соединений диаграмма имеет следующий вид (рис 1.1):
реагентов, абсцисса – координата реакции; 1 – p-комплекс субстрата и реагента; 2 – s-аддукт;
1 2 3 Субстрат и реагент через небольшой барьер образуют p-комплекс, при преодолении более высокого энергетического барьера – s-аддукт и далее через один или два барьера (два – с получением преддиссоциативного p-ком-плекса) – конечный продукт. В субстрате реакции чаще всего имеется несколько атомов, на которые может быть направлена атака реагента. Для образования связи необходимо электростатическое притяжение реакционных центров, т. е. реагирующие атомы субстрата и реагента должны иметь различные по знаку заряды. Кроме того, на занятой молекулярной орбитали (ЗМО) электронодонорной молекулы необходимо иметь пару электронов, которая способна при сближении частиц взаимодействовать со свободной молекулярной орбиталью (СМО) (дыркой) электроноакцепторной молекулы. Рассмотрим реакцию между электронодонорной молекулой S и электроноакцепторной молекулой T. ЗМО молекулы S, содержащая пару электронов, взаимодействует со СМО молекулы T, в результате чего возникает новая связь:
S + T = X
Реакция протекает при следующих условиях: на атоме s, который является реакционным центром электронодонорной молекулы S, имеется пара электронов, отрицательный или частично отрицательный заряд и относительно большой коэффициент атомной орбитали (АО) высшей занятой молекулярной орбитали (ВЗМО). На реакционном центре t электроноакцепторной молекулы T имеется положительный или частично положительный заряд и относительно высокое значение коэффициента АО низшей свободной молекулярной орбитали (НСМО). ВЗМО и НСМО называются граничными орбиталями. По общей многоэлектронной теории возмущений (метод ВМО) изменение полной энергии DЕполн при частичном образовании связи между атомами s и t молекул S и T в растворителе с эффективной диэлектрической константой e описывается следующим фундаментальным уравнением (уравнение Клопмана)
где qs и qt – полные заряды на атомах s и t; Rst – расстояние между атомами s и t, для которых вычисляется энергия; Csm – коэффициенты АО ЗМО – m, атома s молекулы S; Ctn – коэффициенты АО СМО – n, атома t молекулы T; Db – изменение резонансного интеграла при взаимодействии орбиталей атомов s и t на расстоянии Rst; E m * и E n * – энергии граничных орбиталей молекул S и T. При малом радиусе реагирующих частиц (величина Rst мала) и большом заряде на реакционных центрах второй член уравнения (ковалентный член) вносит малый вклад в изменение энергии и им можно пренебречь. В этом случае реализуется зарядовый контроль протекания реакции. Если величина заряда на реагирующих атомах мала и разница между энергиями ВЗМО одной молекулы и НСМО другой незначительна, электростатический член уравнения имеет малое значение. Наибольший вклад в изменение энергии вносит ковалентный член уравнения – орбитальный контроль протекания реакций. По эмпирическому принципу жестких и мягких кислот и оснований (ЖМКО) Пирсона реагенты разделяются на три группы. Жесткие реагенты имеют малый радиус и высокий заряд, мягкие – большой радиус и невысокий заряд. Для промежуточных величин радиуса и заряда реагенты занимают среднее положение. Энергетически выгодно взаимодействие жестких реагентов с жесткими, а мягких с мягкими. Анализ этой ситуации с помощью уравнения Клопмана показывает, что в первом случае наблюдается зарядовый контроль, а во втором – орбитальный. Позднее в качестве меры абсолютной жесткости (h) был предложен количественный параметр, который равен половине разности потенциала ионизации (I) и сродства к электрону (А). В терминах теории МО величине I соответствует ЕВЗМО, а величине А – ЕНСМО. Таким образом, величина абсолютной жесткости может быть найдена по (1.2):
h = ЕВЗМО - ЕНСМО /2 (1.2)
В табл.1.2 приведены некоторые примеры кислот и оснований. Таблица 1.2 Классификация кислот и оснований
*В скобках приведена величина абсолютной жесткости h в эВ
Из данных таблицы видно, что протон является жестким реагентом и его присоединение будет происходить к атомам, имеющим наибольший отрицательный заряд. В реакциях электрофильного замещения в ароматическом ряду участвуют жесткие реагенты, такие как NO2+, SO3, RSO3+, RCO+, Cl+. Для данных реакций характерен зарядовый контроль. Поведение реагентов в реакциях нуклеофильного замещения подробно рассмотрено в разделе 2.2. В качестве примера можно рассмотреть реакции нуклеофильного замещения в молекуле, содержащей мягкий центр – атом хлора и жесткий центр – атом фтора. С жестким основанием, каким является этилат анион, замещается фтор, с мягким реагентом, каким является CH3SNa, – атом хлора:
Приведенная классификация реагентов может быть полезна при разработке оптимального метода синтеза веществ. Ее использование позволяет выбрать субстраты, с помощью которых возможно региоселективное проведение процесса.
Классификация реакций В рассмотренных примерах наблюдается гетеролитический тип образования связи. Одна из классификаций реакций основывается на природе реагента – реакции электрофильного и нуклеофильного замещения. По механизму превращения различают также реакции присоединения реагентов к молекуле или отщепления заместителя от молекулы. Если реагирующие частицы содержат по одному неспаренному электрону, тип образования связи гомолитический (радикальные реакции). В особую группу выделяют перициклические реакции с циклическим переносом электронов, при котором нет заряженных или свободнорадикальных частиц. Примером служит реакция Дильса – Альдера. Различают по механизму также мономолекулярные и бимолекулярные реакции. Если, например, лимитирующей стадией процесса является разрыв связи в субстрате реакции и быстрое взаимодействие образовавшегося иона с другой молекулой, то реакции называют мономолекулярными. Если в лимитирующей стадии процесса участвуют две молекулы – бимолекулярными. По принятой номенклатуре реакции кратко обозначают с помощью букв и цифр, учитывающих тип реакции, реагент и число участвующих молекул. Первая буква – тип реакции: S (substitution) – замещение, A (addition) – присоединение, E (elimination) – отщепление. Вторым подстрочным символом обозначают тип реагента – N (nucleophilic), E (electrophilic), R (radical) – нуклеофильный, электрофильный и радикальный. Третий символ – цифра: 1 – мономолекулярные реакции, 2 – бимолекулярные. Например SN1, SR2, SE2. Специфику реакций ароматических соединений обозначают – Ar. Так реакции электрофильного замещения в ароматическом цикле обозначают SE2Ar или более кратко – SEAr. Реакции, использующиеся в промышленном органическом синтезе, как правило, хорошо изучены. Это можно продемонстрировать на примере реакций электрофильного замещения в ряду ароматических и гетероароматических соединений SEAr.
Механизм и кинетика реакций Реакции электрофильного ароматического замещения имеют общий механизм, который можно представить следующей схемой:
p-комплекс s- аддукт
где реагент Х+ – электрофильная частица, имеющая целочисленный или частично положительный заряд, бензольный цикл является обобщенной формулой ароматического соединения; k 1 – константа скорости прямой реакции образования s-аддукта, k - 1 – обратной реакции, k 2 – константа скорости образования целевого продукта, k -2 – обратной реакции,:В – основание, связывающее уходящий протон.
Для понимания закономерностей реакций, а также для организации технологического процесса полезно использовать формальный кинетический анализ. Рассмотрим общее кинетическое уравнение. Скорость реакции – увеличение концентрации конечного продукта во времени, прямо пропорциональна константе скорости реакции – k 2, концентрации s-аддукта и концентрации агента, связывающего уходящий протон:
V=
Накопления s-аддукта в реакционной массе не наблюдается. Поэтому текущую, стационарную концентрацию этого продукта можно вычислить, приравняв ее приход расходу. Скорость образования промежуточного продукта:
скорость расхода промежуточного продукта:
k 1 [ArH] [X+] = (k 2 [B] + k- 1) [ArHX+] (1.6) Из уравнения (1.6) можно вычислить текущую концентрацию s-аддукта:
Зная формулу расчета концентрации s-аддукта, выраженную через экспериментально определяемые величины, можно рассчитать скорость реакции:
Анализ уравнения (1.8) довольно сложен. Необходимо определить или знать лимитирующую стадию реакции. Во многих случаях (вариант 1) величина k 2[B] существенно больше, чем k- 1 и этим слагаемым можно пренебречь. Уравнение существенно упрощается:
V = k1 [ArH][X+] (1.9)
Поэтому главными являются константа скорости реакции образования s -аддукта и концентрация электрофильной частицы. Энергетическая диаграмма реакции, графически отображающая этот вариант, приведена в начале раздела. Во втором случае лимитирующей стадией процесса является отрыв протона. Тогда k -1 >> k2 [B] и последним слагаемым знаменателя можно пренебречь. Кинетическое уравнение имеет следующий вид:
В энергетической диаграмме реакции этого случая барьер отщепления протона выше, чем барьер образования s-аддукта. Отнесение процесса к первому или второму варианту может быть осуществлено с помощью измерения кинетического изотопного эффекта. Кинетическим изотопным эффектом (КИЭ) называют отношение константы скорости реакции частицы, содержащей легкий изотоп, к константе скорости реакции частицы, содержащей тяжелый изотоп. Величина КИЭ определяется тем, что прочность связи между атомами зависит от массы соответствующего атома. В органическом синтезе обычно имеют дело с отщеплением изотопов атома водорода – протоном и дейтероном. Энергия разрыва связи C-D выше энергии разрыва связи C-H, поэтому дейтерозамещенные соединения реагируют медленнее. При соотношении k H/ k D большем единицы, т. е. когда наблюдается положительный КИЭ, стадия отрыва протона является лимитирующей. Если кинетический изотопный эффект отсутствует, то лимитирующей стадией процесса служит образование s-аддукта. Влияние заместителей может быть оценено с помощью уравнений Гаммета и Тафта.
1.4.4. Уравнения Гаммета, Тафта
Для количественной оценки влияния заместителей Гамметом на основании принципа линейности свободных энергий для ионизации замещенных бензойных кислот была найдена следующая зависимость:
s x = lg k – lg k 0 = - р К а + (р К а)0 (1.11)
где Х – заместитель в п - или м -положении, s - константа заместителя, учитывающая как индуктивный, так и мезомерный эффекты радикала Х, k 0 – константа равновесия для бензойной кислоты (Х = Н), k – константа равновесия для замещенной бензойной кислоты (Х ≠ Н),. Следует отметить, что линейной зависимости для заместителей в о- положении безойной кислоты не наблюдается. Константы некоторых заместителей приведены в табл. 1.3 Таблица 1.3 s-Константы заместителей
Известно, что для бензойной кислоты величина р К а = 4,21. Из уравнения 1.11 могут быть найдены величины р К а замещенных бензойных кислот. Так для п -нитробензойной кислоты р К а = 3,43; м – 3,50; для п- метоксибензойной кислоты р К а = 4,48, а для м- производного р К а = 4,09. В последнем примере четко просматривается влияние как мезомерного, так и индуктивного эффектов. Между заместителем и карбоксигруппой в м -метоксибензойной кислоте нет прямого полярного сопряжения и проявляется только индуктивный эффект, метоксигруппа является электроноакцепторной. В п -метоксибензойной кислоте уже есть сопряжение – метоксигруппа является электронодонором, кислота более слабая. Уравнение Гаммета может быть распространено и на химические реакции и в обобщенном виде может быть представлено в следующем виде
lg k/k 0 = rs, (1.12)
где k – константа скорости реакции для замещенных производных (Х ≠ Н), k 0 – константа скорости реакции для Х = Н, r – константа реакционной серии. Уравнение (1.12) применимо для щелочного гидролиза этилбензоатов (85 % водный этанол, 25 оС). Этилбензоат и замещенные в цикле этиловые эфиры бензойной кислоты называют реакционной серией. Тогда вместо константы равновесия используется константа скорости реакции. Использование данного уравнения полезно для оценки скорости реакции производных, а также для выбора условий проведения процесса. В тех случаях, когда в реакционном центре возникает целочисленный положительный заряд, используют s + константу. При возникновении отрицательного заряда применяют s - константу. Для алифатических соединений Тафтом было разработано аналогичное уравнение, учитывающее стерический эффект заместителя (Es). Для ионизации производных уксусной кислоты найдены s* константы, в которых главную роль играет индуктивный эффект:
lg k/k 0 = rs * + Еs. (1.13)
В отличие от уравнения Гаммета, в уравнении Тафта s * Н = + 0,49, а величина s * для метильной группы равна нулю. Важную роль для оценки влияния заместителей играет и константа r. Знак константы указывает на то, как влияют электронодонорные и электроноакцепторные заместители на константу скорости реакции. Для реакций нуклеофильного замещения в ароматическом ряду величина r имеет положительное значение, для реакций электрофильного замещения знак константы r имеет отрицательное значение, для свободнорадикальных и перициклических реакций величина r близка к единице. Абсолютное значение величины константы r также дает важную информацию химику. Так при реакции этилирования бензола и его производных (C2H5Br, GaBr3, C2H4Cl2, 25 oC) величина r = - 2,4, при нитровании (CH3COONO2, (CH3CO)2O, 25 oC) r = - 6,0, ацетилировании (CH3COCl, AlCl3, 25 оC) значение r составляет - 9,1, бромировании (Br2, CH3COОН, 25 oC) r = - 12,1. При расчете константы скорости реакции для замещенного бензола по уравнению Гаммета очевидно, что влияние заместителя в случае этилирования, в отличие от бромирования, незначительно. Чем меньше абсолютное значение константы r, тем меньше региоселективность реакции. Это можно продемонстрировать на примере хлорирования толуола в различных условиях. Реагент NaOCl, электрофильная частица Cl+, r = -6,0; при хлорировании хлором в уксусной кислоте r = -9,5; при хлорировании хлором в нитрометане r = -13 (температура 25 оС для всех изучаемых реакций). В первом случае выход о - и п -изомеров составляет лишь 95 %, а м- изомера – 5 %, во втором м- изомер образуется с выходом только 0,6 %, а в условиях хлорирования в нитрометане его выход не более 0,1 %. Таким образом меняя условия проведения процесса можно существенно увеличить его региоселективность. Изменение знака r при смене растворителя свидетельствует об изменении механизма реакции. Так при гидролизе арилсульфохлоридов до сульфокислот в воде величина r составляет – 0,30 (механизм реакции SN1), в водном ацетоне r = + 0,84 и механизм реакции становится уже SN2. Влияние растворителя будет подробно рассмотрено в следующих разделах. Полезен также анализ соотношения kп и kм для реакций электрофильного замещения: lg kп/k 0 = s+ n r, а lg kм / k 0 = s м r, соотношение kп/k 0 = nf, соотношение kм/k 0 = mf, nf и mf – парциальные скорости реакции в п- и м- поло-жения цикла. Таким образом, lg n f - lg m f = lg n f / m f = r(s+ п - s м). Соотношение n f и m f позволяет оценить преимущественное направление реакции – вступление заместителя в о-, п- или м- положение цикла. Если принять величину k 0 равной единице, то для замещенного соединения константу скорости реакции называют относительной (k отн), и ее величину можно найти экспериментально. Для этого используют метод конкурирующих реакций – проводят в одном реакционном сосуде взаимодействие реагента с эквимолекулярными количествами бензола и изучаемого производного бензола. Так были проведены реакции бензола и толуола, бензола и хлорбензола с нитрующей смесью, выделены продукты реакции и определены их выходы. Соотношение выхода нитротолуолов и нитробензола было 24,1: 1 – это и есть k отн. Во втором случае соотношение выхода суммы изомерных нитрохлорбензолов и нитробензола составляло 3,3 × 10-2. Для многих реакционных серий эти данные является справочными. По величине k отн можно прогнозировать условия проведения реакции. Так для хлорбензола необходимо использовать более жесткие условия, чем для бензола, например повышение температуры реакции на 20 – 30 оС. Анализ реакционной способности с помощью уравнения Гаммета и его модификаций для производных бензола и гетероциклических соединений не всегда дает надежные результаты. Для практической деятельности полезно использовать справочные данные по величинам r и s при выборе условий проведения реакций в ряду, где уже исследованы некоторые из производных, а также для повышения региоселективности процесса. При разработке плана синтеза важной задачей является определение места вступления заместителя в молекулу по одному из нескольких реакционных центров. Вследствие разнонаправленного действия заместителей и (или) гетероатомов исходя из рассмотрения индуктивного и мезомерного эффекта не всегда удается разрешить поставленную задачу. Ее можно решить с помощью использования квантово-химических расчетов. Тем более что в настоящее время имеются доступные пакеты прикладных программ для их выполнения – MOPAC и HYPERCHEM.
Квантово-химические расчеты
Используются две группы методов:неэмпирические– ab initio и полуэмпирические. Наиболее точные результаты дает ab initio. Оба используют SCF MO self-consistent-field molecular orbitals, т. е. метод самосогласованного поля и приближения молекулярных орбиталей. В неэмпирических методах вычисляют большое число молекулярных интегралов. С увеличением размера молекулы базис расчета возрастает приблизительно пропорционально n4. Величина n – общее число базисных атомных орбиталей (АО). При учете конфигурационных взаимодействий, электронной корреляции, поправок на релятивистские эффекты и неадиабатичность существенно увеличивается время расчета. В простейшем случае неэмпирические расчеты строятся с использованием минимального базиса АО. Каждая из АО в разложении представлена одной орбиталью слейтеровского типа (STO). Минимальный базис, содержащий N гауссовских функций (GTO), примененный для аппроксимации одной слейтеровской, равняется трем – STO-3G. Из-за больших затрат времени на расчеты обычно используют полуэмпирические методы. Разница в том, что в полуэмпирических методах рассматривают только взаимодействия валентных электронов, при этом некоторая часть этих интегралов параметризуется и/или задается константами (именно это и является причиной потери точности), тогда как в ab initio все эти взаимодействия просчитываются (отсюда и большие затраты времени). Приближения, используемые в полуэмпирических методах: - CNDO complete neglect of differential overlap (Pople, Santry, Segal/1965)рассматривает только валентные электроны. Метод аппроксимирован на атомных потенциалах и сродстве к электрону, причем некоторые из них брались как полуэмпирические величины. Первый метод назывался CNDO/1; в настоящее время используется CNDO/2, в нем улучшена параметризация. - MINDO/1 (Barid, Dewar/1969) – первая версия модифицированного INDO. Параметры выбирались на основе экспериментальных молекулярных теплот образования, но метод плохо воспроизводил геометрию молекулы. Поэтому в 1970 г. был улучшен в методе MINDO/2 (дает хорошие результаты по длинам связей и теплотам образования, но плохие по длинам связей с водородом). Следующие версии были MINDO/2’ и MINDO/3. Наиболее удачен последний, его ошибки составили 11 ккал/моль по теплоте образования, 0,02 Å по длинам связей, 5° по углам, 0,4D по дипольному моменту и 0,8 эВ по величинам потенциалов ионизации. - MNDO (модифицированный MNDO) разработан в 1977 г. Его ошибки по величинам: по теплоте образования 9 ккал/моль, 0,025 Å по длинам связей, 3° по углам, 0,36 D по дипольному моменту и 0,5 эВ по потенциалу ионизации. - AM1 (Austin Model 1) разработан на основе MNDO в 1985 г. В нем была улучшена способность воспроизведения водородных связей, по сравнению с предшествующей версией. - PM/3 (MNDO parametric method 3) (первый и второй были MNDO и AM1) существенно снизил ошибки MNDO и AM1, при этом точность расчетов теплот образования, геометрии, дипольного момента не теряется. Сравнение методов MNDO, AM1, РМ3 показывает то, что абсолютная ошибка по Hf298 составляет 22,5 %, 13,8 % и 8,2 % соответственно. В тонком органическом синтезе для оценки направления атаки реагента на один из альтернативных реакционных центров используют величины зарядов на них (зарядовый контроль реакции), величины и знаки коэффициентов атомной орбитали (АО) высшей занятой молекулярной орбитали или низшей свободной орбитали – АОВЗМО или АОНСМО (орбитальный контроль реакции). Для сравнения реакционной способности в ряду соединений важно знать и величины энергии граничных орбиталей ЕВЗМО и ЕНСМО. Эти данные могут быть получены с помощью квантово-химических расчетов. В реакциях электрофильного и нуклеофильного замещения в ряду ароматических и гетероароматических соединений наблюдается зарядовый контроль. В качестве примера на схеме приведены величины зарядов на атомах двух молекул (расчет проведен в приближении АМ1).
Дипольные моменты: 1,615D 5,351D ЕВЗМО = - 9,296 эВ, ЕНСМО = 0,134 эВ ЕВЗМО = - 10,314 эВ, ЕНСМО = -1,263 эВ
Экспериментально найденные величины дипольных моментов данных молекул в газовой фазе составляют соответственно 1,74D и 4,35D, что близко к расчетным данным и подтверждает корректность полученных величин. Для сравнения дипольные моменты молекул, найденные расчетом по методу CNDO/2, составляют 3,151D и 6,72D. Сравнение расчетных величин дипольных моментов, полученных с помощью методов АМ1 и CNDO/2, с экспериментальными позволяет отдать предпочтение методу АМ1. Из величин зарядов на атомах углерода п -хлортолуола видно, что в реакциях электрофильного замещения атака электрофильного агента предпочтительнее в положение 2, хотя не исключается атака и в положение 3. Подробно поведение этого соединения в реакции нитрования будет рассмотрено в разделе 2.1.1. В случае о- нитрохлорбезола атака электрофильного агента предпочтительнее в положение четыре ароматического цикла. Действительно, в случае нитрования о- нитрохлорбезола в основном образуется 2,4-динитрохлорбензол. Если рассмотреть поведение этих соединений в реакциях нуклеофильного замещения, то из данных расчета видно, что при взаимодействии с нуклеофилами замещение атома хлора в о- нитрохлорбезоле возможно, а в п -хлортолуоле проблематично. Таким образом, в сложных случаях с помощью квантово-химических расчетов можно прогнозировать поведение молекулы в различных реакциях. Однако корректность расчетов должна быть подтверждена соответствием полученных результатов с физическими параметрами. Например, с геометрией молекулы, найденной с помощью рентгеноструктурного анализа (РСА), а в приведенных примерах с величинами дипольных моментов молекул. В противном случае из результатов квантово-химических расчетов можно сделать неправильные выводы. Квантово-химические расчеты используют также для описания механизма реакции. При этом они являются единственным некосвенным источником информации о структуре и энергетике переходных состояний, которые в принципе невозможно наблюдать экспериментально. Они также незаменимы при рассмотрении высоколабильных интермедиатов, поскольку экспериментальные методы не позволяют их охарактеризовать. В таких случаях ведут расчет поверхности потенциальной энергии (ППЭ).
Выбор растворителя
В квантово-химических расчетах рассматривается изолированная молекула в газовой фазе. Однако практически все реакции в органическом синтезе осуществляются в растворителях. Даже в тех случаях, когда реакции проводят в расплаве, один из реагентов выполняет функцию растворителя. При проведение процессов в жидкой фазе необходимо рассматривать не только два реагента – субстрат и реагент, но и учитывать влияние третьего компонента – растворителя. В ходе реакции в растворе возникает многокомпонентная система: реагент, субстрат, продукты основной и побочных реакций и растворитель. Специфичная сольватация каждого из компонентов может существенно изменить скорость основной и побочных реакций, а также и селективность взаимодействия. Сольватацию называют неспецифической, если растворитель с веществом дают сольват только за счет электростатического взаимодействия. Специфической сольватацией называют взаимодействие реагента и растворителя с образованием донорно-акцепторных комплексов с переносом заряда или с образованием водородной связи. Следует отметить, что возникновение даже слабой водородной связи может кардинально изменить распределение электронной плотности в молекуле. В ряде случаев взаимодействие между реагентами и растворителем настолько велико, что способствует гетеролитическому разрыву ковалентной связи на ионы. Первоначально ионы находятся в сольватной оболочке – своеобразной клетке из растворителя. За счет диффузии они попадают в одну клетку и реагируют между собой. В качестве растворителей в органическом синтезе используют около 60 жидкостей из примерно 300 жидких веществ органической и неорганической природы. Органических растворителей около 50. Из физических характеристик, определяющих выбор растворителей, следует обращать внимание на диэлектрическую проницаемость e (величина, показывающая степень умень-шения силы электростатического взаимодействия частиц в растворителе по сравнению с вакуумом), полярность и температуру кипения. По величине e растворители условно разбиты на три группы: высокополярные (e > 50), средние (e = 12 – 50) и малополярные (e < 12). Разницу в энергии кулоновского взаимодействия в средах с различными величинами e приближенно можно описать уравнением 1.14:
где m и e – дипольный момент и диэлектрическая проницаемость первого и второго растворителя, r – расстояние между центрами зарядов в полярной молекуле. Энергии активации реакции в разных средах связаны между собой уравнением (1.15). Из уравнений (1.14) и (1.15) следует, что энергия активации реакции полярного вещества в полярном растворителе ниже, чем в неполярном. Величины Ткипиe растворителей,а также их выбор для проведения различных реакций приведены в табл.1.4. Новым направлением для экстракции веществ является использование растворителей, находящихся в сверхкритическом состоянии. Жидкости при температуре и давлении, превышающие критические величины, переходят в состояние, которое не является ни жидкостью, ни газом. Расстояния между присутствующими в сверхкритической фазе частицами (молекулами и кластерами) намного больше, чем в жидкости, но меньше, чем в газе. Сверхкритическая (СК) среда отличается исключительно низкой вязкостью и высокой диффузионной способностью. Наиболее передовыми технологиями являются декофеинизация зерен кофе без размола с помощью СК-СО2 (Ткрит составляет 31,1 оС и Ркрит 7,28 МПа) и очистка смазочных масел от тяжелых фракций с помощью СК-С3Н8 (Ткрит = 96,7 оС и Ркрит 4,19 МПа). При понижении давления ниже критического значения растворитель переходит в газ, а растворенные вещества отделяются. Использование сверхкритических жидкостей в химии и технологии может привести к принципиально новым технологическим решениям.
Таблица 1.4
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-04-08; просмотров: 688; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.227.46.202 (0.018 с.) |

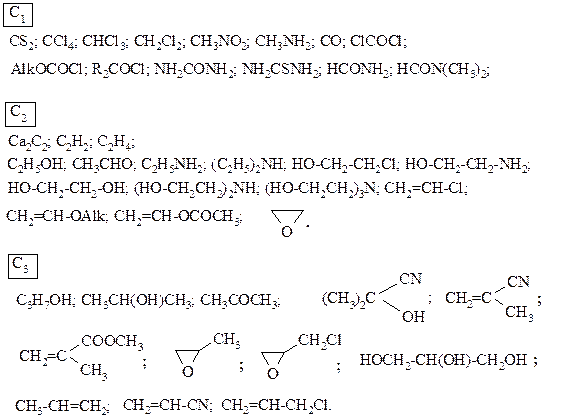









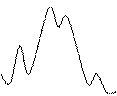 DG Рис. 1.1. Энергетическая диаграмма реакции:
DG Рис. 1.1. Энергетическая диаграмма реакции:
 ордината – изменение свободной энергии системы
ордината – изменение свободной энергии системы
 3 – второй p-комплекс
3 – второй p-комплекс




 = k2 [Ar HX+][B] (1.3)
= k2 [Ar HX+][B] (1.3)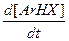 = k 1 [ArH] [X+] (1.4);
= k 1 [ArH] [X+] (1.4); = k 2[ArHX+][B] + k -1 [ArHX+] (1.5)
= k 2[ArHX+][B] + k -1 [ArHX+] (1.5) (1.7)
(1.7) (1.8)
(1.8) (1.10)
(1.10)
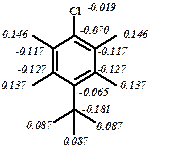

 (1.14)
(1.14) (1.15)
(1.15)


