
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Клинические проявления анемииСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Больных беспокоит одышка, сердцебиение, слабость, могут быть головокружения и обморочные состояния, снижение аппе- тита. При осмотре отмечается бледность кожи и слизистых обо- лочек, восковидный цвет ушных раковин, ломкость волос, может наблюдаться атрофия сосочков языка – «полированный язык». При обследовании выявляется тахикардия, приглушенность I то- на, систолический шум на верхушке сердца, снижение амплиту- ды зубца Т и интервала S-T на электрокардиограмме. Постгеморрагические анемии
Выделяют острые и хронические постгеморрагические ане- мии. Этиология, патогенез и изменения в картине крови при ост- рой постгеморрагической анемии описаны выше в разделе «Па- тология общего объема крови». Хроническая кровпотеря приво- дит к развитию железодефицитной анемии и будет описана в со- ответствующе разделе.
Анемии вследствие недостаточности эритропоэза:
Группа анемий, объединенных одним общим механизмом развития, который связан с нарушением или полным прекраще- нием эритропоэза в результате дефицита веществ, необходимых для осуществления нормального кроветворения, носит название дефицитных анемий. Сюда относят дефицит железа и микро- элементов (медь, кобальт), витаминов (B12, фолиевая кислота В6, В2, С, пантотеновая кислота, никотиновая кислота) и белков.
Классификация анемий вследствие недостаточности эритропоэза:
Железодефицитные анемии Порфиринодефицитные анемии Витамин В12-дефицитные анемии Фолиеводефицитные и др. анемии Гипо-, а- и метапластические анемии Анемии при хронических заболеваниях
Анемии, обусловленные дефицитом факторов образования эритроцитов
Железодефицитные анемии
Наиболее часто встречаются железодефицитные анемии, особенно у детей раннего возраста, девочек в период полового созревания и женщин в бериод беременности и лактации. Данный вид составляет 2/3 случаев от числа всех анемий. Причиной железодефицитных анемий является превышение потерь железа над его поступлением в организм. Суточная по- требность в железе для мужчин составляет 10 мг, для женщин – 18 мг, в период беременности и лактации – 33-38 мг. В норме всасывается только около 10 % (1,5-2 мг в сутки) железа, посту- пающего с пищей, при его дефиците всасывание железа может возрастать до 40 %. Столько же железа теряется с мочой, калом, слущивающимся эпителием и его производными (ногти, волосы), менструальной кровью, грудным молоком. Общая потеря состав- ляет 1,5-2 мг/сут. В 1 мл крови содержится 0,5 мг железа, запасы
Обмен железа в организме изображен на рисунке 14.
Рис. 14. Обмен железа в организме
Всасывание железа лучше происходит из мяса, в меньшей степени – из других продуктов. Неорганическое железо овощей и зерновых в основном трехвалентное, до 60% железа находится в трудноусвояемой форме, связанной с фитиновой кислотой. В же- лудке Fe+3 под действием соляной кислоты желудка превращается в Fe+2, в таком виде железо всасывается быстрее. Ускоряют его всасывание аскорбиновая кислота, ионы меди. Препятствуют всасыванию железа фосфаты, оксалаты, препараты кальция и со- держащие кальций продукты (творог, молоко), а также вегетари- анская диета. Основное влияние на процесс всасывания железа оказывают общие запасы железа в организме, а также степень ак- тивности эритропоэза. В сосудистом русле железо соединяется с трансферрином – гликопротеидом, синтезируемым в печени. Ос- новным источником сывороточного пула железа являются мак- рофаги печени и селезенки, где происходит распад старых эрит- роцитов. При абсорбции его в тонком кишечнике в плазму поступает небольшое количество железа. Большинство клеток, в том числе эритробласты и гепатоциты, содержат на мембране рецепторы к трасферрину. В клетке железо включается в состав ферментов, содержащих и несодержащих гем (табл. 8). Основными формами депонированного железа являются ферритин и гемосидерин, ко- торые содержатся практически во всех тканях организма.
Таблица 8. Гемовое и негемовое железо
Дефицит железа в организме возникает при его недостаточ-ном поступлении с пищей (наиболее богаты железом печень, яй- ца, овсяная, пшеничная, гречневая крупа, мясо, яблоки, черная смородина), при нарушении всасывания (гастриты, энтериты, ре- зекция части желудка и двенадцатиперстной кишки), транспортажелеза кровью (дефицит трансферрина), а также при повышениипотребности в нем (многоплодие, растущий организм, беремен- ность, лактация). Ребенку, рожденному в срок, требуется около 160 мг железа в день, недоношенному, лишенному депо железа – 240 мг (приложение 1), рис 15-16.
К отрицательному балансу железа в организме также могут привести избыточные ежесуточные в течение месяца потери кро- ви в количестве 25-50 мл из ЖКТ (язвенная болезнь, диафраг- мальная грыжа, гастриты: алкогольные или вследствие лечения салицилатами, стероидами, индометацином, неспецифический язвенный колит, болезнь Крона, дивертикулез, варикозно расши- ренные вены пищевода, геморроидальные вены, гельминтоз: по- ражение власоглавом и анкилостомой), гиповитаминоз К (гемор- рагическая болезнь новорожденных и др.). Дефицит железа развивается, когда его потеря превышает 2 мг/сут. У женщин частыми причинами дефицита железа могут быть метроррагии, у мужчин – кровотечения из мочеполового тракта. При метроррагиях женщины могут терять до 40 мг железа за цикл. Повышенный расход железа отмечается в ходе беременно-
Рис. 16. Механизмы развития железодефицитных анемий
Потери железа могут происходить при внутрисосудистом гемолизе вследствие гемоглобинурии и гемосидеринурии при хронических гемолитических анемиях. Недостаток железа в организме приводит к снижению его содержания в плазме крови – сидеропения (в норме 12-30 мкмоль/л), что, в первую очередь, приводит к недостаточному образованию гемоглобина (в составе гемоглобина содержится 1,7-2,8 г железа) и ведѐт к гипохромии эритроцитов. В связи с тем, что железо входит в состав железосодержащих ферментов (цитохромов, пероксидаз, каталазы), при железодефицитных ане- миях нарушаются окислительно-восстановительные процессы в тканях, снижается антиоксидантная защита, что способствует ак- тивации окислительного повреждения клеточных мембран и кле- точному повреждению. Поэтому, кроме общеанемических симптомов при железо- дефицитной анемии развивается сидеропенический синдром, ко- торый проявляется мышечной слабостью, истончением и выпа- дением волос, ломкостью ногтей (койлонихия), трофическими изменениями кожи и слизистой желудочно-кишечного тракта (глоссит, гингивит, атрофический гастрит), изъязвлением в углах рта (ангулярный стоматит), нарушением аппетита, а также из- вращением вкуса и обоняния (употребление в пищу мела, глины, угля, сырых продуктов), дисфагией, диспепсией и снижением иммунитета. У девушек в период полового созревания (ранняя форма) сидеропенический синдром получил название хлороз (от греч. – зеленый), «бледная немочь», у женщин в климактериче- ском периоде отмечается поздняя его форма. В периферической крови при железодефицитных анемиях отмечается понижение содержания эритроцитов, уменьшение уровня гемоглобина, значительное снижение цветового показате- ля (гипохромная анемия), вследствие чего эритроциты выглядят в виде колец. Наблюдается пойкилоцитоз и анизоцитоз с преобла- данием микроцитоза (рис.17). Причиной образования микроцитов является дополнительное митотическое деление предшественни- ков эритроцитов при их созревании в красном костном мозге. Из- вестно, что гемоглобин выступает как ингибитор деления нормо- цитов, а его недостаточное образование способствует дополни- тельному делению предшественников эритроцитов. Количество ретикулоцитов снижается (гипорегенераторная анемия).
Рис. 17. Картина периферической крови с железодефицитной анемией (гипохромия, микроцитоз, анизоцитоз эритроцитов)
В последние годы для характеристики железодефицитной анемии применяют определение в сыворотке крови концентрации растворимых рецепторов к трансферрину, отражающих адекват- ное поступление железа в клетки эритропоэза. При железодефи- цитной анемии происходит повышение синтеза и экспрессии данных рецепторов и увеличение их концентрации в крови. Выделяют три последовательных этапа железодефицитной анемии – предлатентный и латентный дефицит железа и собст- венно железодефицитную анемию (табл. 9). Предлатентный дефицит железа – состояние, предшест- вующее дефициту железа. Клинические симптомы отсутствуют. Уровень гемоглобина нормальный. Показатели транспортного фонда железа в норме. Снижены показатели запасов железа. Латентный дефицит железа сопровождается сидеропени- ческим синдромом, обусловленным дефицитом железа в тканях. Анемия отсутствует, содержание гемоглобина нормальное. Кри- териями диагностики латентного дефицита железа являются из- менение показателей транспортного фонда железа (снижение же- леза сыворотки, повышение общей и латентной железосвязы- вающей способности сыворотки, нормальные или сниженные значения коэффициента насыщения трансферрина), снижение уровня ферритина сыворотки. Диагностика собственно железодефицитной анемии осно- вывается на выявлении признаков анемии в общем анализе крови, морфологического исследования мазка периферической крови. Снижение уровня гемоглобина следует считать проявлением же- лезодефицитной анемии только в том числе, если выявляются ги- похромия (морфологически и по цветовым индексам), понижен- ный уровень ретикулоцитов и снижение транспортного фонда железа. Биохимически отмечается уменьшение содержания железа в сыворотке до 1,8-7,2 мкмоль/л (сидеропения) и увеличение общей железосвязывающей способности (в норме 40,6-62,5 мкм/л) как следствие компенсаторного повышения концентрации трансфер- рина). Процент насыщения трансферрина снижается до 15 % и ниже.
Таблица 9. Лабораторные критерии железодефицитных со- стояний у детей
Железоперераспределительные анемии
Второе место по распространенности среди всех анемий за- нимают железоперераспределительныеанемии, сопровождающие хронические инфекционные, ревматические, опухолевые заболе- вания и обусловлены нарушением перемещения железа из депо в плазму. Для нормального гемопоэза организму в сутки требуется 25 мг железа. В тонком кисшечнике всасывается 1,5-2 мг железа. Основная доля железа поступает в клетки гемопоэза путем его реутилизации из разрушающихся эритроцитов. В ряде случаев клетки депо (макрофаги селезенки, печени, костного мозга) прочно удерживают железо, поэтому механизм реутилизации на- рушается. Развивается перераспределительный, или функцио- нальный, дефицит железа, сопровождающийся снижением дос- тавки железа к эритробластам костного мозга, нарушению эри- тропоэза и развитию анемии. Предполагается, что активация макрофагов при хрониче- ских воспалительных заболеваниях приводит к продукции про- воспалительных цитокинов, которые способны повышать синтез ферритина и подавлять экспрессию гена эритропоэтина. В периферической крови отмечается умеренная гипохром- ная анемия, умеренное снижение гемоглобина, снижение сыворо- точного железа, ОЖСС, трансферина, коэффициент «насыщение трансферрина железом» (НТЖ) и повышение содержания сыво- роточного ферритина.
Порфиринодефицитные анемии
Сходными по механизму развития с железодефицитными анемиями являются порфиринодефицитные (сидероахрестиче- ские, греч. achrestos – бесполезный, тщетный ) анемии. Они раз- виваются при нарушении включения железа в гем из-за низкой активности ферментов, участвующих в синтезе порфиринов, ко- торые входят в состав гема. Порфирины также являются обяза- тельными компонентом каталазы, пероксидаз, цитохромов, а также миоглобина. Порфиринодефицитные анемии могут быть наследственными (сцепленные с Х-хромосомой или аутосомой) и приобретенными (интоксикация свинцом, дефицит витамина В6). Нарушение включения железа в гем приводит к увеличению уровня железа в плазме крови (до 80-100 мкмоль/л) и накоплению его в печени, надпочечниках, поджелудочной железе, яичках, что нарушает их функции. НТЖ достигает 100%.
Лечение железодефицитных анемий должно быть направле- но на устранение нарушения поступления и всасывания железа в организм, лечебное питание, восполнение депо железа и прове- дение противорецидивной (поддерживающей) уровень железа в организме терапии. Среди препаратов железа лучше всего ис- пользовать сульфат железа (по 200 мг три раза в сутки перед едой). При железоперераспределительных анемиях препараты железа не назначаются.
Витамин В12-дефицитная анемия
Особое место занимает анемия, связанная с дефицитом ви- тамина В12 (суточная потребность около 1 мкг, в дневной рацион – 10-15 мкг), относящаяся к анемиям, возникающим вследствие дефицита кровеобразующих факторов. Витамин В12-дефицитная анемия (болезнь Аддисона- Бирмера, пернициозная, злокачественная) впервые описана Ад- дисоном (1849 г.), затем Бирмером (1872 г.). В продуктах питания цианокобаламин, или «внешний фактор Кастла» связан с белком. В желудке витамин В12 освобождается от белка и связывает- ся с синтезируемым париетальными клетками гликопротеином – «внутренним фактором Кастла», что позволяет образовавшемуся комплексу всасываться в тонкой кишке. В крови витамин соеди- няется с транспортными белками траскобаламинами (рис. 17). Большая часть витамина связывается с транскобаламином I, в соединении с которым он неактивен, а также связывается с транскобаламином II, который с током крови транспортирует ви- тамин в печень, костный мозг, головной мозг и другие органы и с транскобаламином III, функция которого неизвестна. Запасы витамина В12 в организме находятся в печени, со- ставляют около 2-5 мг. При прекращении экзогенного поступле- ния цианокобаламина их достаточно на 3-6 лет. Основными причинами В12-дефицитных анемий являются нарушение поступления витамина в организм вследствие вегета- рианского питания и рождения от матерей с дефицитом витамина В12; нарушение усвоения витамина В12 в организме вследствие рака и хронических атрофических заболеваниях желудка и тонко- го кишечника, состояниях после резекции данных органов, ток- сического действия высоких доз этанола на слизистую желудка, врожденного нарушения продукции «внутреннего фактора Каст- ла», «конкурентного» использования цианокобаламина паразита- ми (широкий лентец, власоглав) и микроорганизмами; нарушение транспорта витамина В12 из-за снижения образования транскоба- ламина, или появления антител к нему; повышенный расход ви- тамина вследствие многоплодной беременности, гемолитической анемии, злокачественных новообразований; снижение запасов витамина В12 вследствие цирроза печени; нарушения усвоения витамина костным мозгом (эритромиелоз).
Витамин В12 имеет две коферментные формы: метилкобала- мин и 5-дезоксиаденозилкобаламин. Метилкобаламин участвует в обеспечении нормального (эритробластического) кроветворе- ния в результате образования из фолиевой кислоты (фолата) тет- рагидрофолиевой (тетрагидрофолата), необходимой для образо- вания тимидинмонофосфата, синтеза глутаминовой кислоты, пу- риновых и пиримидиновых оснований. Тимидинмонофосфат включается в ДНК эритрокариоцитов, обеспечивая нормобласти- ческий тип кроветворения в красном костном мозге, а также в ДНК других быстропролиферирующих клеток (эпителий кишеч- ника, семенников). Другая коферментная форма 5-дезоксиаденозилкобаламин регулирует синтез жирных кислот, катализируя превращение ме- тилмалоновой кислоты в янтарную (сукциниловую) кислоту, ко- торая необходима для образования миелина. Недостаток витамина В12 приводит к нарушению синтеза и структуры ДНК, что уменьшает количество митозов в эритропо- эзе (рис.18). В результате клетки эритроидного ростка увеличи- ваются в размерах (мегалобласты и мегалоциты), резко уменьша- ется их количество и продолжительность жизни из-за низкой ре- зистентности (рис.19).

Рис. 18. Нормальный (эритробластический тип кроветворения (справа) и патологический (мегалобластический) – слева. Рис. 19. Картина крови при В12- и фолиеводефицитной анемии Одновременно нарушается созревание клеток других рост- ков гемопоэза (мегакариоцитарного, миелоцитарного, лимфоци- тарного), что приводит к тромбоцитопении и лейкопении. При В12-дефицитной анемии в результате тяжелого функ- ционального поражения костного мозга вследствие авитаминоза В12 регенерация эритроцитов протекает по мегалобластическому типу кроветворения (рис.18). При этом родоначальником новооб- разованных эритроцитов является мегалобласт, который при дальнейшем созревании превращается в зрелую безъядерную клетку – мегалоцит. Мегалобласты и мегалоциты обнаруживают- ся при В12-дефицитной анемии не только в костном мозге, но и в крови. Нормоциты, наоборот, встречаются в крови при анемии Бирмера исключительно редко. Мегалобласты представляют собой крупные клетки в 2-4 раза больше эритробласта. Цитоплазма их нередко гиперхромна, ядро – относительно большое с нежной сетевидной структурой, бедное хроматином. При созревании мегалобласта ядро не вы- талкивается, в отличие от нормобласта, а распадается на фраг- менты, часть которых выталкивается из клетки, а часть лизирует- ся. Поэтому в цитоплазме мегалобластов часто видны тельца Жолли (остатки ядер) и остатки нуклеолеммы (кольца Кебота). Тельца Жолли представляют собой точечные включения в прото- плазме эритроцита, окрашивающиеся по Романовскому-Гимза в яркокрасный цвет, обычно их не более 1-2 в эритроците. Кольца Кебота представляют тонкие колечки или фигуры в виде вось- мерки и окрашиваются по методу Романовского-Гимза в красный или синий цвет. Характерно ассинхронное созревание ядра и ци- топлазмы. После исчезновения ядра из мегалобласта образуется мега- лоцит. Мегалоцит отличается от нормоцита большей величиной (диаметр его равен 12-20 мкм) и более интенсивной окраской (гиперхромия), что соответствует высокому цветовому показате- лю крови. Гиперхромия отличает мегалоциты от макроцита. Витамин В12-дефицитные анемии отличаются очень низким содержанием эритроцитов (1,0-0,8 × 1012 /л) и гемоглобина (50-25 г/л). Цветовой показатель превышает норму, достигая 1,4-1,8 (ги- перхромная анемия). В периферической крови характерно нали- чие мегалобластов и мегалоцитов, в эритроцитах телец Жоли и колец Кебота, анизоцитоза и пойкилоцитоза, лейкопении, трмбо- цитопении. Отмечается наличие нейтрофилов с пиперсегменти- рованным ядром. Количество ретикулоцитов обычно понижено. Вследствие нарушения регенераторной способности костно- го мозга при витамин В12-дефицитных анемиях развиваются из- менения со стороны желудочно-кишечного тракта (глоссит и формирование «полированного» языка вследствие атрофии его сосочков (атрофический глоссит Гунтера, стоматит, гастроэнте- роколит), которые обусловлены нарушением деления и созрева- ния эпителиоцитов слизистой (рис. 19). Для дефицита витамина В12 характерно также наличие нев- рологического синдрома, вызванного нарушением образования миелина дорзальных и латеральных канатиков спинного мозга, включающего признаки фуникулярного миелоза (шаткая поход- ка, парестезии, болевые ощущения); психические расстройства (бред, галлюцинации). Неврологический синдром обусловлен де- фицитом 5-дезоксиаденозилкобаламина, накоплением метилма- лоновой кислоты и недостатком янтарной кислоты. Лечение анемий состоит в назначении внутримышечных инъекций этого витамина (по 1 мг, инъекции повторяют каждые 2-3 дня, пока не будет сделано 6 инъекций). Затем проводят по одной инъекции раз в три месяца, пока не будет устранена при- чина дефицита витамина В12.
Фолиеводефицитные анемии
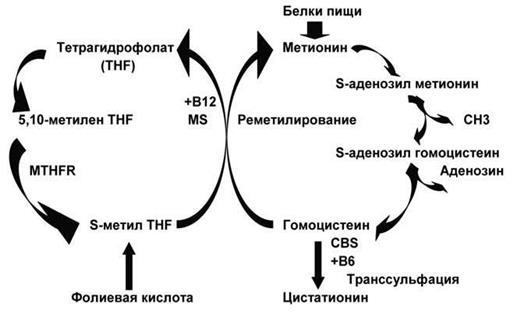
Соединения фолиевой кислоты (фолаты, фолацин) содер- жатся в большом количестве в печени, мясе, дрожжах, шпинате. Суточная потребность взрослого человека в фолиевой кислоте составляет 100-400 мкг. При дефиците поступления запасы ее в организме (5-20 мг) исчерпываются в течение 3-4 мес. Всасыва- ется фолиевая кислота в основном в верхнем отделе тонкой киш- ки. Коферментная форма фолиевой кислоты – тетрагидрофо- лиевая – необходима для образования тимидинмонофосфата, синтеза глутаминовой кислоты, пуриновых и пиримидиновых ос- нований, участвует в нейрофизиологии мозга. Причинами дефицита фолиевой кислоты может быть: пло- хое питание (диета, бедная зеленью, свежими фруктами, овоща- ми), нарушение всасывания фолацина (кишечная мальабсорбция, алкоголизм, прием барбитуратов, гастрэктомия), повышенная по- требность в фолиевой кислоте (беременность, кормление грудью, болезни кожи, гемобластозы), блокирование перехода фолиевой кислота в тетрагидрофолат под влиянием аминоптерина, метот- рексата (ингибиторы дигидрофолатредуктазы). Дефицит фолие- вой кислоты приводит к аналогичным изменениям в крови и же- лудочно-кишечном тракте, которые наблюдаются при витамин В12-дефицитной анемии. Установлено, что витамины В12 и фолиевая кислота участ- вуют в реметилировании гомоцистеина в аминокислоту метионин (рис.20). Дефицит этих витаминов приводит к увеличению кон- центрации гомоцистеина в крови. Гипергомоцистеинемия вызы- вает повреждение и активацию эндотелиальных клеток, что спо- собствует развитию атеросклероза, инфаркта миокарда, тромбоза.
Рис. 20. Метаболизм гомоцистеина. MTHFR – метилентетрагидрофолат-редуктаза, MS – метионин-синтаза, CBS – цистатионин-β-синтаза
Лечение дефицита фолиевой кислоты состоит в назначении фолиевой кислоты (5 мг/день на протяжении 4-х месяцев, затем режим изменяют – либо 5 мг фолиевой ксилоты раз в неделю, ли- бо 400 мкг – ежедневно).
Развитие мегалобластических анемий возможно не только по причине дефицита витамина В12 и (или) фолиевой кислоты, но также в результате нарушения синтеза пуриновых или пирими- диновых оснований, необходимых для синтеза нуклеиновых ки- слот. Причиной этих анемий обычно является наследуемое (как правило, рецессивно) нарушение активности ферментов, необхо- димых для синтеза фолиевой, оротовой, адениловой, гуаниловой и, возможно, некоторых других кислот. В результате этого нару- шаются структура ДНК и заключенная в ней информация по син- тезу полипептидов, что ведет к трансформации нормобластиче- ского типа эритропоэза в мегалобластический. Проявления ука- занных анемий в большинстве своем такие же, как при витамин В12-дефицитной анемии.
Другие виды дефицитных анемий
В основе развития анемий может быть также дефицит меди, цинка (входит в состав карбоангидразы) и др. Медь – второй после железа по значению гемопоэтический микроэлемент. Медь (кофактор полифенолоксидазы, тирозиназы, цитохромоксидазы, лактазы, моноаминооксидазы) необходима для эритропоэза и, вообще, – миелопоэза. Участвует в стимуля- ции созревания ретикулоцитов путем активации цитохромокси- дазы, а также модуляции захвата железа трансферрином, что не- обходимо для включения железа в гем, ускоряя синтез гемогло- бина и участвуя в синтезе железопорфиринов. В развитии эритрона принимают участие никель, молибден, которые входят в состав ферментов пуринового обмена, марганец – в состав амино-ацил-т-РНК-синтетаз, селен – в состав антиок- сидантной системы. Важное значение в развитии эритрона имеют незаменимые аминокислоты (гистидин, изолейцин, триптофан, лизин). При их дефиците нарушается эритропоэз.
Витамин В2 участвует в функционировании эритроцитар- ной глютатионредуктазы, предохраняющей эритроциты от ауто- окисления. Арибофлавиноз проявляется анемией, особенно ха- рактерной для недоношенных и грудных детей.
Витамин В6 используется как кофактор дегидрогеназы δ- аминолевулиновой кислоты, участвующей в начальных этапах синтеза гема, а также некоторых других ферментах порфорино- вого обмена. При алкоголизме из-за ускорения разрушения вита- мина В6 вследсвие действия ацетальдегида на активность фер- мента оксидазы отмечается нарушение утилизации железа кост- ным мозгом, сидеробластическая анемия, гемохроматоз.
Витамин H, как кофермент карбоксилаз, а также витамин С – компонент редокс-системы глутатиона влияют на резистент- ность эритроидных клеток к аутоокислению и при их дефиците может наблюдаться анемия смешанного патогенеза. Витамин E участвует в сдерживании процесса эритродиэре- за, вызываемого активными кислородными радикалами.
Гипо-, а- и метапластические анемии
Анемии, вызванные нарушением образования эритроцитов вследствие преимущественного повреждения либо стволовых клеток либо клеток-предшественниц миелопоэза и/или эритропо- этинчувствительных клеток, называются гипо- или апластиче- скими анемиями. Наследственные апластические анемии:
(Блекфена-Даймонда).
Гипо- и апластические анемии развиваются вследствие по- вреждения стволовых клеток и подавления миелопоэза в костном мозге либо из-за наследственных дефектов гемопоэтических кле- ток (первичные) либо при воздействии на костный мозг физиче- ских, химических, биологических факторов (вторичные, или при- оберетенные анемии). При наследственной апластической анемии (анемия Фан- кони), характеризующейся врожденными соматическими анома- лиями, наблюдается дефект гемопоэтических клеток, наследуе- мый по аутосомно-рецессивному механизму, вследствие которого нарушаются процессы репарации ДНК. Физические (ионизирующее излучение), химические (лево- мицетин, бутадион, цитостатики, бензол, мышьяковистые соеди- нения, инсектициды), биологические (вирусы, антиэритроцитар- ные антитела и цитотоксические Т-лимфоциты) факторы вызы- вают гипоплазию костного мозга, нарушают синтез ДНК, белка в стволовых клетках вследствие изменения физико-химического микроокружения стволовых клеток либо отмены иммунологиче- ской толерантности. Вследствие нарушения пролиферация стволовых клеток и образования зрелых форменных элементов периферической кро- ви развивается панцитопения. Содержание гемоглобина в эрит- роцитах близко к норме (нормохромная анемия), однако общее содержание Hb резко снижено (до 20-30 г/л). Количество ретику- лоцитов снижено или они отсутствуют (гипо- или арегенератор- ная анемия). В настоящее время термин апластическая анемия употребляется для панцитопений со сниженной функциональной способностью гипопластического, замещенного жиром костного мозга. При этом выявляются следующие синдромы, связанные с недостаточностью миелопоэза:
 анемический синдром (количество гемоглобина падает до 20-30 г/л), анемический синдром (количество гемоглобина падает до 20-30 г/л),
гемолитический синдром, геморрагический синдром, инфекционно-септический синдром. Клинически выявляются признаки гипоксии (недомогание, слабость, гиподинамия, головная боль, быстрая утомляемость), тромбоцитопения и снижение свертываемости крови (кровоиз- лияния), лейкопения (склонность к развитию инфекционных про- цессов). У младенцев может наблюдаться приобретенная транзитор- ная апластическая анемия с парциальным поражением эритропо- эза.
Анемии, развивающиеся при вытеснении клеток нормально- го эритропоэза опухолевыми вследствие лейкозов либо метаста- зов в костный мозг, носят название метастатических. Анемии при хронических заболеваниях
Анемии при хронических заболеваниях характеризуются неспособностью костного мозга гиперплазироваться и увеличи- вать эритроидную массу в достаточной мере соответственно сте- пени анемии. Основными причинами являются хроническая по- чечная недостаточность, гипотиреоз, пангипопитуитаризм, бел- ковое голодание. В патогенезе анемии при хронической почечной недоста- точности имеет значение дефицит эритропоэтина, укорочение продолжительности жизни эритроцитов, токсическое влияние на эритроциты продуктов азотистого обмена, кровопотери, обуслов- ленные дефектом тромбоцитов. Важную роль в развитии нефро- генной анемии играют ингибиторы эритропоэтина – ФНО-α, ИЛ- 1, γ-интерферонов, секретируемые активированными макрофага- ми. В периферической крови выявляется нормохромная, реже ги- похромная нормоцитарная анемия. Количество ретикулоцитов обычно нормальное или незначительно снижено.
Гемолитические анемии
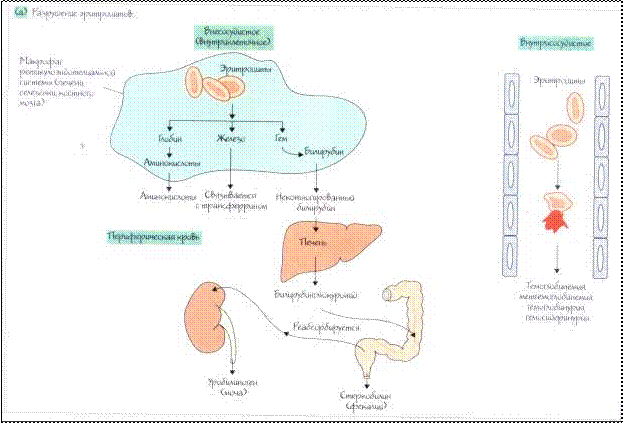
Большую группу анемий составляют гемолитические ане- мии (ГА). Главным патогенетическим фактором в возникновении этого вида анемий является укорочение срока жизни эритроцитов и преобладание процесса разрушения эритроцитов над их про- дукцией. При развитии гемолитической анемии продолжитель- ность жизни эритроцитов сокращается до 15 дней и меньше. Ге- молитические анемии чаще встречаются в детском возрасте. В нормальном организме постоянно происходит физиологи- ческое разрушение эритроцитов, вследствие их естественного старения. Продолжительность жизни эритроцитов составляет в среднем 120 дней. Основная масса эритроцитов (90 %) разруша- ется путем фагоцитоза макрофагами селезенки, печени и красно- го костного мозга (внутриклеточный гемолиз), рис. 21–22. При этом гемоглобин подвергается превращению в билирубин, а за- тем в уробилин и стеркобилин и выводится с мочой и калом. Ос- тальная часть эритроцитов (10 %) распадается непосредственно в кровотоке (внутрисосудистый гемолиз). Освобожденный гемо-
Рис. 21. Разрушение эритроцитов
Преждевременное разрушение эритроцитов осуществляют преимущественно клетки системы мононуклеарных фагоцитов (внутриклеточный гемолиз). В некоторых случаях лизис эритро- цитов усиливается в сосудистом компартменте (внутрисосуди- стый гемолиз). При большинстве приобретенных гемолитических анемий имеет место усиленный внутрисосудистый гемолиз вследствие действия экзоэритроцитарных факторов, тогда как на- следственные формы чаще имеют усиленный внутриклеточный гемолиз, обусловленный эндоэритроцитарными причинами. При активации внутрисосудистого гемолиза в крови увели- чивается количество свободного гемоглобина выше 0,04 г/л или 4 мг%. При этом гемоглобин быстро связывается с β2- глобулиновым белком гаптоглобином, образуя комплекс, что приводит к снижению уровня гаптоглобина в сыворотке крови
Рис. 22. Мононуклерно-фагоцитарная система
При внутрисосудистом гемолизе может повышаться уровень метальбумина, соединения, образованного свободными группами гема из метгемоглобиновых комплексов и альбумином. При патологическом внутрисосудистом гемолизе гаптогло- бинсвязывающая способность истощается, и гемоглобин выделя- ется через почки с мочой, окрашивая ее в красно-коричневый цвет. Реабсорбированный гемоглобин, превращаясь в гемосиде- рин, вызывает гемосидероз почечных канальцев. Вследствие инициирования трехвалентным железом свободно-радикального окисления развивается некробиоз канальциевого эпителия и его слущивание в мочу, страдает функция почек. При внутриклеточном гемолизе возрастает уровень пре- имущественно неконьюгированного билирубина (> 17 мкмоль/л), стеркобилина кала и уробилина мочи. При этом гемолитические анемии сопровождаются желтухой, т.е. характерным окрашива- нием склер, слизистых, кожи пациента вследствие избытка в кро- ви и отложения в тканях непрямого билирубина (при превыше- нии уровня общего билирубина 85,5 мкмоль/л). Избыток билиру- бина экскретируется ЖКТ в виде стеркобилина, придающего фе- калиям темный цвет. Повышается вероятность образования в желчном пузыре и желчных протоках пигментных камней (холе- литиаз).
|


 железа в организме составляют 3-4 г.
железа в организме составляют 3-4 г. Рис. 15. Обмен железа и этиопатогенез железодефицитных анемий
Рис. 15. Обмен железа и этиопатогенез железодефицитных анемий сти, родов (потеря 150-200 мг железа) и лактации (900 мг).
сти, родов (потеря 150-200 мг железа) и лактации (900 мг).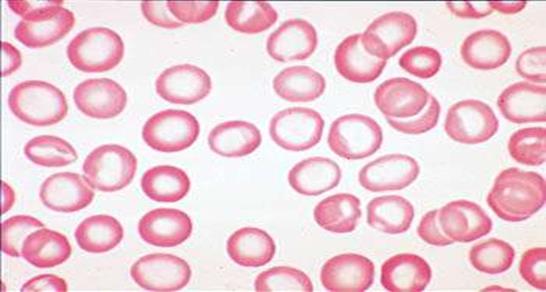
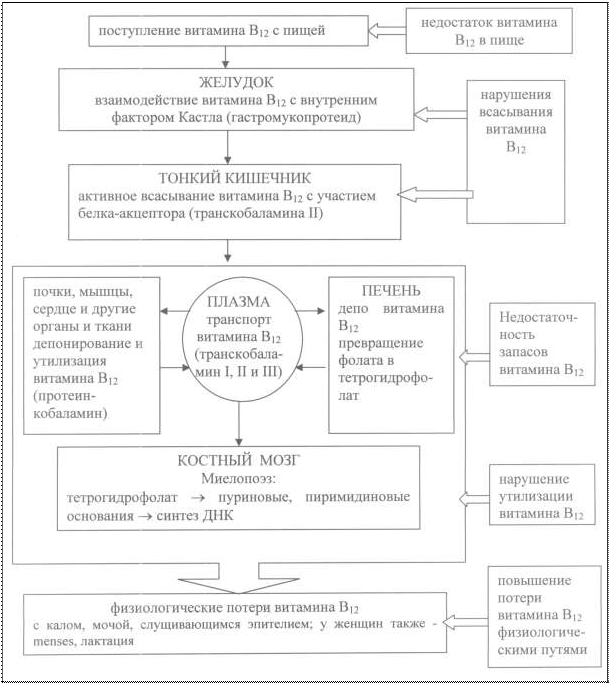 Рис. 17. Обмен витамина В12 и патогенез В12-дефицитных анемий
Рис. 17. Обмен витамина В12 и патогенез В12-дефицитных анемий

 глобин при этом связывается с белком плазмы – гаптоглобином, а затем этот комплекс разрушается макрофагами печени и селезен- ки.
глобин при этом связывается с белком плазмы – гаптоглобином, а затем этот комплекс разрушается макрофагами печени и селезен- ки. (гипогаптоглобинемия). Гемоглобин, несвязанный с гаптоглоби- ном, превращается в метгемоглобин.
(гипогаптоглобинемия). Гемоглобин, несвязанный с гаптоглоби- ном, превращается в метгемоглобин.


