Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Общие тесты свертывания кровиСодержание книги
Поиск на нашем сайте
Наиболее распространенные тесты (время свертывания цельной крови, время рекальцификации плазмы, толерантность плазмы к гепарину) позволяют судить лишь об общем состоянии гемостаза. Длительность времени свертывания крови (ВС) по Ли—Уайту находится в пределах 5—10 мин, нормальное физиологическое значение толерантности плазмы к гепарину (ТП) по Сиггу — 9—13 мин, времени рекальцификации плазмы (ВР) по Бергерхофу — Рокку 90—120 с. Укорочение времени каждого из этих тестов свидетельствует о гиперкоагуляции, удлинение— о гипокоагуляции. Тестами тромбоцитарной функции являются: 1. Время кровотечения (ВК). Нормальное время кровотечения по Дькжу составляет 1—3 мин. Увеличение ВК наблюдается при болезни Виллебранда, тромбоцитопении, тромбоцитопатиях, после приема ацетилсалициловой кислоты. ВК в определенной степени отражает также функцию и контрактильную способность сосудистой стенки. 2. Число тромбоцитов. Нормальное содержание тромбоцитов 150—400-109/л. Тромбоцитопения может считаться непосредственной причиной кровотечения при снижении числа тромбоцитов до 50-109/л и ниже. 3. Агрегация тромбоцитов. Если к плазме, обогащенной тромбоцитами и хорошо перемешанной, добавить АДФ, то тромбоциты начинают образовывать агрегаты и конгломераты. При постоянной концентрации АДФ выраженность агрегаций зависит от числа тромбоцитов, способности их к агрегации, наличия плазменных факторов коагуляции, в частности фибриногена. Таким образом, при перечисленных условиях тест позволяет получить представление об агрегационных способностях тромбоцитов и, следовательно, оценить степень возможного участия качества тромбоцитов в процессе гемостаза и при гиперкоагуляционном состоянии. 4. Фактор 3 тромбоцитов. Тест потребления протромбина (см. ниже) наиболее прост для оценки активности фактора 3. Нормальные тромбоциты вызывают постепенное укорочение времени свертывания, которое колеблется между 20 и 40 мин.
Тесты на активность факторов коагуляции: 1. Протромбиновое время (тест Квика) (ПВ). В норме составляет 12—14 с. Продолжительность теста 15 с принимается за 100% (протромбиновый индекс). Тест позволяет оценить активность факторов внешнего механизма свертывания крови. Укорочение ПВ (увеличение протромбинового индекса) свидетельствует об усилении тромбообразования в крови и увеличении тромбогенной опасности. Гепарин, поскольку он дает главным образом антитромбиновый эффект, обесценивает протромбиновый тест. Показатель теста можно считать условно достоверным только через 5 ч после последнего введения гепарина. Использование тканевых прокоагулянтов при выполнении протромбинового теста нивелирует роль факторов VIII, IX, XI и тромбоцитов в ходе коагуляционного процесса. В связи с этим протромбиновый тест в таких условиях дает возможность оценить дефицит факторов II, V, VII, X и фибриногена. Заболевания печени, дефицит витамина К проявляются удлинением ПВ. Наблюдается также увеличение ПВ в поздних стадиях ДВС-синдрома. ПВ является наиболее точным методом контроля эффективности терапии антикоагулянтами непрямого действия.
2. Активированное частичное тромбопластиновое время (АЧТВ). Представляет собой тест, отражающий совокупность активности всех факторов внутреннего механизма свертывания крови. Определяемое по Раппопорту АЧТВ в норме составляет 22—40 с. Укорочение АЧТВ является признаком усиления тромбопластической активности крови и повышенного темпа образования тромбина в крови. Удлинение АЧТВ наблюдается при гемофилии, циррозе печени, применении антикоагулянтов прямого действия, а также ДВС-синдроме, сопровождаемом «потреблением» факторов коагуляции крови. Хотя тест АЧТВ чувствителен к дефицитам всех факторов коагуляции, за исключением фактора VII, обычно он используется для того, чтобы определить степень участия в процессах коагуляции первых стадий механизма коагуляции, т. е. тех, которые контролируются факторами XII, XI, IX и VII. Как видно из изложенного, протромбиновое время по Квику не отражает дефектов коагуляции в I стадии (период генерации тромбопластина), определяемых дефицитом факторов внутреннего механизма. Тест АЧТВ, выполненный в сочетании с ПВ, позволяет отличить дефекты коагуляции в I стадии от дефектов коагуляции во II стадии (образование тромбина), когда проявляют активность факторы II, X, V и когда проявляется уже развившаяся активность фактора VII, или в III стадии, когда из фибриногена образуется фибрин. Удлинение АЧТВ при нормальном ПВ неоспоримо указывает на дефицит одного из факторов в I стадии.
3. Тест потребления протромбина. Превращение протромбина в тромбин (потребление протромбина) является функцией скорости появления в крови протромбинпревращающих факторов во время активного процесса образования сгустка. Основой теста является сравнение содержания протромбина в сыворотке крови через 1 ч после образования сгустка с содержанием протромбина в исходной плазме, из которой получен сгусток. Отклонения от нормы могут отмечаться при состояниях, которые характеризуются дефицитом факторов, ответственных за развитие комплексов, превращающих протромбин в тромбин. Это факторы VIII, IX, X, XI, XII, а также тромбоциты и фактор V. Тест позволяет отличить дефицит всех предшественников в генерации активного фактора X (Ха) и, следовательно, самого фактора X от дефицита фактора VII, поскольку для пробы, осуществляемой in vitro, фактор VII не требуется. Таким образом, у больного с дефицитом фактора VII тест потребления протромбина будет нормальным при удлиненном ПВ. Наоборот, у больного с дефицитом фактора X будет наблюдаться удлиненное ПВ при сниженном тесте потребления протромбина. Тесты, характеризующие фибриноген, фибринолиз и действие гепарина: 1. Тромбиновое время (ТВ) по Сирмаи (норма 25—30 с) может отражать изменения концентрации и структуры фибриногена. Оно может быть увеличено введением гепарина и повышением уровня в крови продуктов деградации фибриногена или фибрина (ПДФ). При потреблении фибриногена в условиях ДВС-синдрома ТВ удлиняется. Протамин не удлиняет ТВ. У больных с острым первичным фибринолизом наблюдается ускоренный лизис сгустка, полученного при постановке теста ТВ. При этом фибриновый сгусток не должен начать растворяться раньше чем через 5 мин. 2. Рептилазное время (РВ). Удлинение РВ (норма 20—22 с) наблюдается при гипофибриногенемии или при увеличении содержания ПДФ, в частности при ДВС-синдроме или вторичном фибринолизе. Поскольку гепарин не влияет на РВ, сравнение последнего с ТВ или ПВ может быть показателем участия гепарина в сдвигах гемокоагуляции. Оценка фибринолиза. 1. Время лизиса эуглобулино-вого сгустка по Ковальскому должно составлять в норме 4—5 ч. Укорочение его до 1—2 ч (вместе с появлением ПДФ) указывает на повышение активности фибринолитической системы. 2. Продукты деградации фибрина (фибриногена) (ПДФ). В норме содержание ПДФ не должно превышать 10 мкг/мл. Повышение концентрации ПДФ всегда указывает на процесс фибринолиза, который может быть первичным, обусловленным повышением уровня плазмина (фибринолизина), или вторичным как результат непрерывного избыточного образования фибрина или его аномальных форм, в частности при ДВС-синдроме. Поздние стадии ДВС-синдрома характеризуются высокой концентрацией ПДФ (свыше 80—100 мкг/мл). 3. Количественное определение плазминогена и плазмина основано на использовании теста на фибриновой пленке и считается чувствительным и точным показателем фибринолиза. Патологический фибринолиз может быть точно диагностирован только в течение 24 ч. В связи с этим в клинической практике метод не имеет практической ценности. Вместе с тем он может быть полезен при оценке эффективности антифибринолитиче-ской терапии с помощью е-аминокапроновой кислоты.
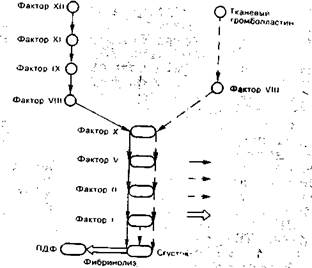
Рис. 6.1. Основные тесты, характеризующие состояние гемостаза. Сплошные стрелки — АЧТВ, пунктирные — протромбиновое время, штрихпунктир-ная — тромбиновое и рептилазное время, светлая — время лизиса эуглобулинового сгустка.
Оценка антитромботической активности. Уровень антитромбина-III ниже 80% свидетельствует о потреблении этого фактора, которое может быть связано с развитием ДВС-синдрома. Это один из самых чувствительных показателей развития внутрисосудистого свертывания крови. К сожалению,, проба на антитромбин-Ш в присутствии гепарина и ПДФ в крови становится невозможной. Концентрация антитромбина-III иногда зависит от гемодилюции, поэтому необходимо трактовать его осторожно и всегда учитывать уровень гематокрита, при котором оценивается этот фактор. Таким образом, описанные тесты (рис. 6.1) могут дифференцированно отражать дефекты гемокоагуляционного процесса и фибринолиза на различных уровнях. Ход процесса свертывания крови и последующего лизиса образующегося сгустка может быть оценен также методом тромбоэластограммы (ТЭГ), получаемой с помощью специального прибора — тромбоэластографа. ТЭГ дает довольно широкое представление о ходе коагуляционного процесса и коагуля-ционном потенциале. Следует помнить, что антикоагулянтная терапия существенно меняет ТЭГ и в ряде наблюдений делает ее результаты недостоверными.
Варианты расстройств системы гемостаза Клиническая ориентировка в диагностике. У большинства больных с врожденными расстройствами гемостаза болезнь проявляется еще в детском возрасте. Обычно после незначительных травм, экстракции зубов, при малых оперативных вмешательствах возникают неостанавливаемые или с трудом останавливаемые кровотечения. При подозрении на врожденные аномалии гемокоагуляции больного необходимо тщательно обследовать, а также провести гематологическое обследование членов семьи. Кровотечение, возникшее вследствие патологического состояния тромбоцитов или их дефицита в крови, может быть остановлено продолжительным давлением в месте повреждения и обычно после этого не возобновляется. В противоположность этому при кровотечениях, обусловленных дефектами коагуляционного процесса, т. е. когда невозможно образование сгустка или замедление его образования и повышен фибринолиз, давление в месте повреждения сосуда эффекта не дает. Прекратившись, такое кровотечение, как правило, вскоре возобновляется (обычно из-за лизиса сгустка). Подобные кровотечения нередко возникают через несколько часов после операции и бывают не слишком интенсивными. Это классический вариант кровотечения, обусловленного расстройствами коагуляции крови. Если не проводится лечение, то такое кровотечение может продолжаться буквально сутками. Другой характерной чертой подобных кровотечений является отсутствие образования сгустка излившейся крови.
Хирургические кровотечения (если у больного коагуляционная система в норме) развиваются весьма драматично, с высокой начальной интенсивностью и никогда не бывают генерализованными, т. е. не сопровождаются кровотечением из мест уколов при инъекциях в других областях тела (если не осложняются ДВС-синдромом). Патология тромбоцитов. Основными лабораторными тестами для оценки роли тромбоцитов в процессе гемокоагуляции являются подсчет тромбоцитов в камерах и определение времени кровотечения. При нормальных показателях этих тестов можно быть уверенным, что кровотечение не связано с расстройством функции тромбоцитов или их дефицитом. Нормальное содержание тромбоцитов в крови 150—400-109/л. Спонтанные кровотечения, обусловленные дефицитом тромбоцитов, возникают тогда, когда количество тромбоцитов существенно снижается и достигает 50—20-109/л. Обычно наблюдаются кровоточивость слизистых оболочек, например рта, десен, петехиаль-ные высыпания в местах давления на кожу, например после наложения жгутов на конечности или измерения артериального давления с помощью манжетки сфигмоманометра. Время кровотечения обычно удлиняется при снижении числа тромбоцитов до уровня ниже 100-109/л. Тромбоцитопения может быть результатом снижения продукции костного мозга, избыточной периферической утилизации, или деструкции клеток, или их активного поглощения увеличенной селезенкой. Продукцию тромбоцитов можно оценить путем подсчета числа мегакариоцитов в пунктате костного мозга. Снижение числа тромбоцитов в сочетании с уменьшением числа мегакариоцитов может свидетельствовать об апластиче-ской анемии или злокачественной инфильтрации костного мозга при лейкемическом или вторичном раковом процессе. Утилизация (потребление) тромбоцитов может иметь; место также при ДВС-синдроме, когда в формирующиеся внутри сосудов тромбы включается большое число тромбоцитов, а костный мозг не успевает их продуцировать. Существуют и другие зоны потребления тромбоцитов, например образование внутрисосудистых гиалиновых тромбов при тромботической тромбо-цитопенической пурпуре или гемолитическом уремическом синдроме. Тромбоцитопения может быть также обусловлена активацией тромбоцитов и их последующей утилизацией в результате дефицита простациклина. Трансфузия плазмы или ее замена может прервать процесс избыточной тромбоцитарной активности, поскольку при этом в кровь поступает достаточное количество плазменных факторов, способствующих высвобождению простациклина из сосудистой стенки {Byrnes J. S., Liam E., 1979]. Иммунные механизмы также играют роль в процессах изменения физиологической активности тромбоцитов и в процессах их потребления. На оболочке тромбоцитов находятся аутоантитела иммуноглобулинов класса G [Идельсон Л. И., 1980]. Это обусловливает их преждевременную деструкцию и фагоцитов преимущественно макрофагами в селезенке и печени. Такие антитела могут быть определены радиоиммунными методами. В большинстве случаев острая иммунная Тромбоцитопения вызывается каким-либо острым заболеванием, например острой бактериальной или вирусной инфекцией, однако может существовать и в хроническом варианте (как идиопатическое заболевание) или сопровождать системную красную волчанку и хроническую лимфоидную лейкемию.
Тромбоцитопения может быть обусловлена и лекарственными веществами. В плазме крови лекарства или их метаболиты могут образовывать комплексы с белками, которые способны проявлять себя как антигены. На поверхности тромбоцитов образуются антитела к иммуноактивным комплексам антигенов. Происходит вторичная адсорбция комплексов на поверхности тромбоцитов, преждевременно разрушающая их. Как известно, нормальная продолжительность жизни тромбоцитов около 10 дней. В случаях иммунных конфликтов продолжительность жизни тромбоцитов укорачивается до нескольких дней, а в большинстве случаев — до нескольких часов. Нормальная селезенка взрослого человека (масса 150— 200 г) способна аккумулировать одновременно около 30%, тромбоцитной массы. В норме эта аккумулированная часть находится в постоянном обмене с массой циркулирующих в крови тромбоцитов. При патологическом увеличении селезенка способна потреблять значительно большее число тромбоцитов, особенно если продукция их в костном мозге повреждена каким-либо патологическим процессом. Возникает Тромбоцитопения. Во всех случаях патологической и необъяснимой кровоточивости кожи или слизистых оболочек следует исключить патологию тромбоцитов, даже если общее число их в периферической крови не изменено. Расстройства функционального состояния тромбоцитов могут быть первичными, связанными с каким-либо изменением качества самих тромбоцитов, например с изменением их метаболизма, или вторичными, возникающими в результате основного заболевания, например сепсиса. Основными функциональными качествами тромбоцитов, как известно, являются их способность прилипания к поврежденной, поверхности, т. е. адгезия, склеивание между собой, т. е. агрегация, выделение образовавшейся массой факторов, которые инициируют процесс коагуляции фибриногена, и веществ, способствующих последующей ретракции образовавшегося сгустка. Следовательно, функции тромбоцитов чрезвычайно многообразны. Расстройства этих функций врожденного или приобретенного характера могут существенно расстроить весь процесс коагуляции крови и гемостаза. Такие расстройства хорошо описаны R. M. Hardisty (1977). Тромбоциты могут приклеиваться к коллагеновой и неколлагеновой поверхности эндотелия. Для адгезии к коллагену кофактор не требуется. При врожденном синдроме Элерса—Данлоса наклонность к кровотечениям связана с тем, что нормальные тромбоциты не могут достаточно прочно соединиться с патологически измененной структурой коллагена [Karaca M. et al.. 1972]. Адгезия тромбоцитов к неколлагеновым структурам зависит от их взаимодействия с двухвалентными катионами (прежде всего Са2+), фибриногеном и фактором Виллебранда. Таким образом, дефекты процесса адгезии тромбоцитов к поврежденной поверхности могут быть связаны с патологией фибриногена, болезнью Виллебранда, гипокальциемией, дефектами самой мембраны тромбоцитов. Описана также группа патологических синдромов, обусловленных дефицитом сиаловой кислоты и гликопротеина I в оболочке тромбоцитов. Эта группа патологических состояний, объединенных общим названием «синдром Бернара — Сулье», характеризуется тромбоцитопенией и потерей адгезивной способности тромбоцитов из-за отсутствия на их оболочке рецепторов фактора Виллебранда. В лабораторных условиях заболевание может быть установлено при выявлении потери тромбоцитами способности адгезироваться на стандартизированной стеклянной поверхности или поврежденной интиме аорты крысы в перфузионной камере. После адгезии коллаген, тромбин и АДФ связываются со специфическими рецепторами на оболочке тромбоцита и таким образом активируют ферментную систему, которая высвобождает свободную арахидоновую кислоту из связанных с мембраной фосфолипидов. Арахидоновая кислота под влиянием цикло-оксигеназы превращается в эндопероксид, который является предшественником тромбоксана А2 и других простагланДинов. Эндопероксид, инициирующий реакцию высвобождения из плотных гранул и тромбоксана А2,— наиболее мощный активатор агрегации тромбоцитов. Эти реакции показаны на схеме 6.3. Возможно, существуют другие механизмы агрегации, которые не зависят от метаболизма арахидоновой кислоты. Это активация коллагеном и большими количествами тромбина, Са2+ и, наконец, тромбоцитоактивирующим фактором [Demopoulos С. А., 1979]. Тромбастения (или болезнь Гланцманна—Негели) представляет собой врожденный дефицит гликопротеина II на мембране тромбоцита, в результате которого нарушается агрегация тромбоцитов при сохраненной способности к адгезии и ристо-цетинобусловленной способности их к агрегации [Баркаган 3. С., 1980]. Врожденный дефицит ферментов циклооксигеназы или тром-боксансинтетазы встречается очень редко, но приобретенный дефицит циклооксигеназы наблюдается чаще. Действие ацетилсалициловой кислоты — его классический пример. Эта кислота необратимо ингибирует циклооксигеназу путем ее ацетилирова-ния [Roth G. J., 1975]. Поскольку циркулирующие тромбоциты не способны самостоятельно синтезировать этот белок, действие разовой дозы ацетилсалициловой кислоты продолжается до полного исчезновения старых тромбоцитов и замены их новыми, т. е. практически до 10 дней. В клинической практике после приема 300 мг ацетилсалициловой кислоты нарушение агрегационной и адгезивной функций тромбоцитов и возможная наклонность к кровотечениям могут поддерживаться 4—7 дней, т. е. в течение периода, необходимого для наработки костным мозгом достаточного количества мегакариоцитов и появления достаточного количества тромбоцитов новой генерации. Некоторые лекарственные вещества, например индометацин и другие нестероидные противовоспалительные средства, ингибируют циклооксигеназу, но кратковременно, и геморрагическая тенденция после их приема может продолжаться не более 24 ч. Агрегационная способность может быть оценена на агрегометре по результатам воздействия индуцирующих агрегацию веществ на плазму, обогащенную тромбоцитами. Известны две группы врожденных дефектов освобождения. Одна из них связана с полным отсутствием плотных гранул и содержащейся в них АДФ, другая — с недостаточностью механизмов освобождения плотных гранул тромбоцитов, несмотря на достаточное количество самих гранул. В клинических условиях кровотечения могут быть связаны с рядом различных отклонений в функциональной активности тромбоцитов. При уремии, например, это чаще всего нарушения адгезивной и агрегационной способности тромбоцитов [Evans E. et al., 1972]. Возможно, что такие расстройства связаны с множеством факторов. Нельзя исключить также влияние различных диализируемых субстанций на функциональную активность поверхности тромбоцитов. В настоящее время известен ряд плазменных факторов, которые стимулируют освобождение простациклина из эндотелия сосудистой стенки и, следовательно, способны ингибировать адгезивную и агрегационную активность тромбоцитов [Remuz-zi G. et al., 1978]. Простациклин ингибирует агрегацию тромбоцитов путем связывания со специфическим мембранным рецептором, который повышает внутриклеточное содержание цАМФ. Последний тормозит синтез тромбоксана А2. В расстройствах тромбоцитарных функций могут играть роль и нарушения миелопролиферативных процессов. Появление тромбоцитов из злокачественно измененного клона мегакариоцитов предполагает возможность нарушений ферментативных функций таких тромбоцитов [Weinfeld A. et al., 1975]. Полицитемия и увеличение эритроцитной массы или тромбоцитоз могут быть причиной парадоксального сочетания тромбозов и кровотечений. При этом повышение вязкости крови ухудшает кровоток и предрасполагает к тромбозам, а дефекты тромбоцитарной мембраны обусловливают геморрагическую тенденцию. У больных с гипергаммаглобулинемией происходит адсорбция гамма-глобулина на поверхности тромбоцитов. Возникает повышенная наклонность тромбоцитов к агрегации и адгезии и следовательно, к тромбозам. Это наблюдается при макроглобулинемии Вальденстрема, множественной миеломе, системной красной волчанке. Известны также нарушения функции тромбоцитов при цинге, пернициозной анемии, болезнях печени (особенно при печеночной недостаточности), клапанных пороках сердца. Описаны расстройства тромбоцитарной функции после переливания низкомолекулярных декстранов и гидроксиэтилкрахмала. Тромбоцитемия начинает клинически проявляться тогда, когда число тромбоцитов превышает 900—700-109/л. Резко возрастает риск тромбозов и тромбоэмболии, особенно артериальных сосудов. Тромбоцитемия может быть первичной, вследствие злокачественной гиперпродукции костным мозгом мегакариоци-тов, или вторичной, как реакция на какие-либо патологические состояния, например обширную травму, оперативное вмешательство, инфаркт миокарда, коллагенозы, лимфоматозы. У ряда больных выраженная тромбоцитемия возникает после спленэктомии. В связи с этим очевидно, что нормализация числа тромбоцитов должна быть достигнута как можно быстрее, желательно путем устранения индуцирующего фактора. Возможны также другие лечебные мероприятия. Производят контролируемую сепарацию тромбоцитов, применяют методы подавления функции костного мозга бисульфаном или подавление функциональной активности тромбоцитов ацетилсалициловой кислотой. Все это существенно снижает риск тромбозов. Тромбоцитопения имеет множество причин. При необходимости лечения тромбоцитопенических кровотечений иногда используют трансфузию цельной свежей крови, однако предпочтительнее трансфузия тромбоцитной массы. Необходимость лечения тромбоцитопении в клинических условиях возникает в тех случаях, когда число тромбоцитов становится меньше 50-109/л. Однако следует помнить о технических трудностях. Это прежде всего быстрая потеря функциональной активности тромбоцитов при их хранении после забора и сепарации. Обычно функциональная активность их остается удовлетворительной не более 48—72 ч. Тромбоциты, хранимые при температуре 4 С, имеют короткие сроки жизни после переливания и эффективны не более 24 ч [Slichter S. J., Marker L. A., 1976]. Их предпочтительнее использовать для лечения острых кровотечений или их последствий. Если тромбоциты хранятся при температуре 22 °С, то продолжительность их жизни после переливания (и эффект) увеличивается примерно до 48—72 ч. Более целесообразно производить их переливание некровоточащим больным с тромбоцитопенией [Valeri С. R., 1976] Нарушения коагуляционной функции. Как уже указывалось, ориентировочную информацию о первичном звене расстройства коагуляции как причины геморрагического синдрома можно получить, применяя скрининговые тесты. Каждый из трех приведенных тестов способен в наибольшей степени отражать узкий диапазон дефекта, поскольку характеризует один из трех механизмов образования сгустка: внутренний, внешний и непосредственный механизм превращения фибриногена в фибрин. Следовательно, специфичность каждого из тестов для отдельных факторов коагуляции можно представить следующим образом (табл. 6.2).
Таблица 6.2. Информативность коагуляционных тестов для отдельных факторов свертывания *
* См. также рис. 6.1.
АЧТВ максимально отражает более ранние стадии образования сгустка, т. е. те, на которых начинается действие факторов внутреннего механизма — XII, XI, IX, наконец, VIII. Наоборот, сдвиги ПВ в большей степени отражают более поздние этапы коагуляции крови — образование фактора Ха, дефицит фактора V (акселератор), недостаток протромбина или дефицит (избыток) фибриногена. Если обнаружено удлинение АЧТВ, подтверждающееся при повторных постановках теста с использованием 50% смеси плазмы больного и нормальной плазмы, то это может свидетельствовать о дефиците какого-либо из названных факторов или их ингибиции, например иммуноглобулином G, который инактивирует один из коагуляционных белков. Дефицит плазменных факторов можно корригировать инфузией нормальной плазмы, тогда как ингибирующее влияние иммуноглобулина корригировать трансфузией плазмы нельзя и удлинение АЧТВ останется прежним. Причины дефицита коагуляционных факторов различны. Он может быть связан с нарушением или полным прекращением синтеза коагуляционных протеинов в печени, качественными нарушениями молекул белков, из которых образуется коагуляционный протеин, или, наконец, с дефицитом ферментов, необходимых для синтеза. Дефицит коагулирующих факторов нередко связан с повышенным потреблением фактора в коагуля-ционном процессе, в частности при ДВС-синдроме, наконец, при инактивации факторов циркулирующими в крови патогенными антителами или ингибиторами [,Machin S. J., 1983]. Все факторы коагуляции крови, кроме VIII (антигемофильный глобулин), синтезируются в печени. Фактор VIII образуется в эндотелиальных клетках как антиген и приобретает коа-гуляционную активность уже в кровотоке. Точная последовательность этого процесса неизвестна, однако предполагается, что происходит образование комплекса с какими-то низкомолекулярными субстанциями [Machin S. J., 1983]. Врожденные расстройства коагуляции. Описаны различные состояния, связанные с дефицитами коагуляци-онных факторов, но все они весьма редки, за исключением гемофилии, болезни Кристмаса (гемофилия В) и болезни Вил-лебранда. Гемофилия и болезнь Кристмаса передаются как сцепленный с полом рецессивный признак. Болезнь Виллебранда обычно обусловлена передачей аутосомного доминантного признака. Все остальные врожденные болезни коагуляции крови являются, как считают, аутосомными рецессивными. Классическая гемофилия является результатом синтеза патологической молекулы фактора VIII, не проявляющей специфической биологической активности, но повышающей уровень иммунологически активных веществ (антиген). Содержание тромбоцитов в крови обычно нормальное, так же как их функция [Bloom A. L., 1977]. Клинически тяжесть заболевания обычно тесно коррелирует с дефицитом нормального фактора VIII. При дефиците его менее 50% признаки болезни, как правило, отсутствуют. При наличии его менее 5% нормы развиваются продолжительные эпизоды кровотечения, которые трудно контролируются. Характерны гемартрозы. При полном отсутствии фактора VIII, если не проводится специальная терапия, очень часты тяжелые спонтанные кровотечения после незначительных повреждений. Иногда возможен смертельный исход. Основой терапии гемофилии (гемофилических кровотечений) является возмещение дефицита фактора VIII донорским препаратом. Благодаря такой профилактической терапии стала возможной нормальная жизнь тяжелобольных. Поскольку продолжительность жизни донорского фактора VIII в крови больного невелика и через 10—14 ч остается половина перелитой дозы, для уверенного контроля в случаях острого кровотечения необходимо двукратное в течение суток введение донорского препарата. Криопреципитат готовят из свежей крови, лиофилизируют и многократно фракционируют для получения концентрата. Приготовленные концентраты чисты, имеют дозированную активность фактора VIII в малом объеме и могут быть легко введены самостоятельно в домашних условиях. Однако при введении антигемофильных препаратов высок риск заболевания вирусным гепатитом, хроническим заболеванием печени, приобретения аллоантител к эритроцитам, тромбоцитам, HLA и плазменным белковым антигенам [McVerry В. A., Machin S. J., 1979]. В последние годы лечение и поддержание больных с гемофилией осложняет проблема СПИДа. Приблизительно у 5—10% больных гемофилией постепенно увеличивается количество ингибиторов фактора VIII, развивается резистентность к лечению криопреципитатами и лиофилизированными концентратами. Эпизоды кровотечений становятся чаще и в конце концов возникает необходимость применения стероидных препаратов, иммуносупрессоров, интенсивного плазмообмена, применения бычьего или свиного фактора VIII или очень высоких доз человеческого фактора VIII [Penner J. A., Kelly P. E., 1975]. Весьма сходна с истинной гемофилией ситуация при болезни Кристмаса (дефицит фактора IX — антигемофильного фактора В), при которой также наблюдается тенденция к спонтанным кровотечениям. Период полураспада фактора IX в циркулирующей крови довольно короткий (до 24 ч), поэтому лечение болезни Кристмаса предпочтительнее проводить с использованием свежезамороженной плазмы и концентратов фактора IX. Болезнь Виллебранда характеризуется продолжительным временем кровотечения, снижением адгезивной способности тромбоцитов, уменьшением содержания коагулоактивного фактора VIII и его иммунной антигенной активности [Meyer D., 1977]. При классической болезни Виллебранда одновременно снижаются все три показателя активности фактора VIII, хотя описано, например, состояние, при котором нормальное содержание антигена сочеталось со снижением адгезивной активности тромбоцитов и изменением их электрофоретической активности. У некоторых больных с синдромом Виллебранда в молекуле фактора VIII уменьшается содержание одного из углеводов, в результате чего тормозятся процессы адгезии и ристоцетиновой агрегации. Некоторые приобретенные формы болезни Виллебранда могут возникать у больных с аутоиммунными заболеваниями [Ingram G. I. et al., 1971]. У них накапливаются антитела, которые преципитируют часть молекулы фактора VIII и нарушают нормальный процесс адгезии тромбоцитов. Обычно наблюдаются лимфоматозы и коллагенозы. Наиболее эффективны при лечении подобных больных криопреципитаты и менее эффективны концентраты фактора VIII. Как уже указывалось, другие врожденные расстройства функции плазменных факторов свертывания крови встречаются реже. Они могут быть достаточно просто выявлены скрининговыми тестами. Обычно их лечение связано с необходимостью возмещения недостающего фактора, и это, как правило, может быть достигнуто инфузией свежезамороженной плазмы. Скри-нинговым методом не удается выявить дефицит фактора XIII (фибринстабилизирующий фермент), поскольку дефицит обнаруживается не ранее 2—3 сут после образования фибринового сгустка. Дефицит фактора XIII диагностируют обычно по ускоренному лизису образовавшегося сгустка в моче. Дефицит витамина К. Жирорастворимый витамин К необходим для синтеза печенью факторов II (Протромбина), VII, IX и X. В связи с этим названные факторы принято именовать витамин-К-зависимыми. Витамин К синтезируется в кишечнике при участии кишечных бактерий. Его абсорбция в кишечнике происходит с участием желчи. Витамин К действует путем кар-боксилирования глутаминовых остатков молекул аминокислот [Brozovic M., 1976]. Механизм действия его заключается в связывании одного из названных факторов с поверхностью фосфолипида в присутствии Са2+. Благодаря этому фактор становится функционально активным и участвует в процессах дальнейшего каскада, т. е. превращается из профермента в фермент. При дефиците и в отсутствие витамина К печень синтезирует неполноценные белки, которые не способны связываться с поверхностью фосфолипида. В печени образуется некоторое количество иммуноактивных аминокислотных соединений, которые сходны с нормальными белками и могут быть выявлены иммунологическими методами. В иностранной литературе эти соединения названы PIVKA (протеины, вызванные отсутствием витамина К или антагонизмом к нему). Сами по себе они также могут несколько ингибировать коагуляционный процесс. Их появление в крови неопровержимо свидетельствует об отсутствии витамина К. Однако при заболеваниях печени, приводящих к нарушению синтеза белков, продукция PIVKA прекращается. Если уровень витамина К в организме снижается, то активность витамин-К-зависимых факторов снижается со скоростью, соответствующей периоду их полураспада в крови (фактора VII—2—4 ч, IX —25 ч, фактора X —40 ч, фактора II —60 ч). У новорожденных в течение первых 3—5 дней имеется дефицит витамин-К-зависимых факторов из-за функциональной незрелости печени и сниженных запасов витамина К (стерильность кишечника и отсутствие витамина К в материнском молоке). Введение ребенку 1 мг витамина K1 полностью подавляет геморрагический синдром. Большие дозы витамина К нежелательны, так как могут вызвать гемолитическую желтуху из-за дефицита гликолитических ферментов. Дефицит витамина К наблюдается у больных с механической желтухой, поскольку желчь у них не попадает в область образования витамина — в кишечник. Среди других причин дефицита витамина К могут быть названы язвенный стоматит, длительная диарея, фиброзный цистит, длительное лечение минеральными маслами (вазелин). Стерилизация кишечника, например длительное применение антибиотиков, также снижает синтез витамина К. Для коррекции этих состояний необходимо внутривенное введение 10 мг витамина K1 ежедневно в течение недели. Длительный прием производных кумарина (пелентан, фенилин) внутрь также ингибирует активирующее действие витамина К на факторы II, VII, IX, X. Поскольку существует множество лекарственных веществ, которые при комбинации с кумариновыми препаратами могут либо усиливать, либо ослаблять их действие, необходим контроль за эффективностью лечения кумаринами с применением ПВ-теста. Если передозировка кумариновых препаратов приводит к ятрогенному кровотечению, то больному необходимо ввести свежезамороженную плазму или концентрат протромбинового комплекса. Болезни печени. Кровотечения часто осложняют заболевания печени. Время жизни гемокоагулирующих фа
|
|||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-30; просмотров: 986; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.51.35 (0.021 с.) |