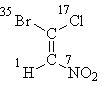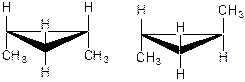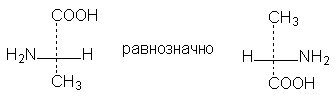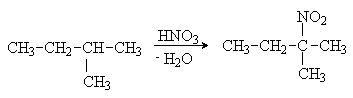Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Классификация органических соединенийСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Лекция № 1 КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ Органические соединения классифицируют по двум основным признакам: строению углеродного скелета и функциональным группам. По строению углеродного скелета различают ациклические (алифатические), циклические (карбоциклические и гетероциклические) соединения. Ациклические соединения – содержат открытую цепь атомов углерода. Карбоциклические соединения – содержат замкнутую цепь углеродных атомов и подразделяются на алициклические и ароматические. К алициклическим относятся все карбоциклические соединения, кроме ароматических. Ароматические соединения содержат циклогексатриеновый фрагмент (бензольное ядро). Гетероциклические соединения - содержат циклы, включающие наряду с атомами углерода один или несколько гетероатомов (О, N,S и др.). По природе функциональных групп органические соединения делят на классы. Например, функциональная группа – гидроксильная –ОН, класс – спирты, карбоксильная –СООН- класс карбоновые кислоты. Соединения близкого строения, но отличающиеся по составу на гомологическую разность (СН2), образуют гомологический ряд. В настоящее время в органической химии общепринятой является систематическая номенклатура (заместительная и радикально-фунукциональная), разработаннаяМеждународным союзом чистой и прикладной химии (IUPAC). Наряду с ней сохранились и используются тривиальная и рациональная номенклатуры. Лекция № 2
Химическая связь Атом углерода имеет s и p – орбитали. Углерод в таблице Д.И.Менделеева занимает шестой порядковый номер; квантовая ячейка невозбужденного атома углерода в основном состоянии имеет следующий вид:1s2 2s2 2p2, возбужденного атома углерода: 1s22s1 2pх12ру1 2рz1. Переход атома углерода в возбужденное состояние возможен в связи с тем, что, во-первых, 2s и 2p- уровни близки между собой энергетически, во-вторых, наиболее низкое энергетическое состояние системы достигается при максимальном числе электронов с параллельными спинами. Таким образом, четырехвалентный атом углерода обладает на внешнем электронном слое набором атомных орбиталей: 2s, 2px, 2pу, 2pz. Углерод в возбужденном состоянии должен образовать ковалентные связи с четырьмя другими атомами. Например, с атомами водорода в молекуле метана, при этом три 2р-орбитали перекрываются с 1s- орбиталями трех атомов водорода, 2s- орбиталь с 1s- орбиталью четвертого атома водорода, следовательно, образуются четыре С-Н связи, отличающиеся по прочности. Однако это противоречит практике, согласно квантово - механическим расчетам все четыре связи равноценны. Для разрешения данной проблемы Л. Полинг сформулировал два постулата: направленной валентности и гибридизации орбиталей. Он предложил, что валентные орбитали (2s, 2px, 2pу, 2рz) гибридизуются (смешиваются) и образуют эквивалентные (одинаковые по форме и энергии) атомные гибридные орбитали. Электронная плотность гибридизованных орбиталей сконцентрирована по одну сторону от ядра, что обеспечивает максимальное перекрывание орбиталей, а значит образование более прочной химической связи. SP3- гибридизация. В образовании гибридной орбитали участвуют одна s- и три р-орбитали. Sp3- гибридизация характерна для насыщенных соединений (углеводородов и их производных). Следует отметить, что распределение электронной плотности sp3 – орбиталей строго направленно и сконцентрировано по одну сторону от ядра, что обеспечивает максимальное перекрывание. Молекула метана СН4 представляет собой идеальный тетраэдр с углом Н-С-Н, равным109028/. Все четыре σ - С-Н равноценны, образованы перекрыванием sp3 - s- орбиталей. В молекуле гомолога – этана СН3-СН3 углерод – углеродная связь образуется перекрыванием двух sp3- орбиталей с образованием σ- связи за счет осевого перекрывания. Длина связи С-С равна 1,54 Ао, прочность σ - связи составляет 350,0 кДж/моль. SP2- гибридизация. В ненасыщенных соединениях атом углерода находится в sp2- гибридном состоянии, т.е. происходит смешение одной s – орбитали с двумя р- орбиталями с образованием трех эквивалентных sp2 - орбиталей. Каждая sp2- орбиталь имеет цилиндрическую симметрию относительно одной из трех осей, расположенных в плоскости под углом 1200. Ось четвертой атомной негибридизованной орбитали расположена под прямым углом к плоскости, в которой лежат три оси sp2-гибридных орбиталей. Перекрывание р-орбиталей (боковое перекрывание) приводит к образованию другого вида ковалентной связи - π. Длина двойной связи составляет 1,34 Ао или 0,134 нм. Электронная плотность π-связи концентрируется выше и ниже плоскости σ- связей в этилене. Поскольку молекула планарна, она легко поляризуется. π- связь менее прочна, чем σ- связь, т.к. р- электронные орбитали с параллельными осями перекрываются значительно меньше, чем при образовании теми же р- электронами или s- электронами σ- связи (перекрывание по оси орбиталей). Общая прочность (σ+π)- связей в этилене составляет 607, 1 кДж/моль. Следовательно, прочность π- связи – 257,1 кДж/моль. sp- гибридизация – комбинация 2s – орбитали и одной 2р - орбитали. При этом образуются две эквивалентные гибридные sp - орбитали, расположенные под углом 1800. У каждого атома углерода остаются по две негибридизованные р- орбитали, которые, перекрываясь, образуют две π- связи. Таким образом, тройная связь между двумя углеродными атомами описывается в рамках теории гибридизации как сочетание одной σ- и двух π- связей. Тройная связь ацетилена менее прочна, чем можно было ожидать из суммы σ- и 2-х π- связей. Известно, что энергия углерод – углеродной связи в этане 350,0 кДж/моль, в этилене – 607,1 кДж/моль, а в ацетилене – 822,7 кДж/моль. Разница энергий связей для ацетилена и этилена 215,6 кДж/моль, т.е. меньше, чем для этилена и этана (257,1 кДж/моль). Длина тройной связи 0,120 нм, т.е. меньше, чем двойной связи (0,135 нм) и тем более простой (0,154 нм). Такое изменение длины связи (С-С) в различных гибридизациях объясняется увеличением доли s- орбитали в гибридной орбитали. Ниже представлна зависимость структуры от гибридизации атома углерода. Гибридизация Геометрия молекулы Примеры sp3 тетраэдрическая Предельные углеводороды, циклоалканы, спирты и др.
sp2 тригональная Этилен и его гомологи, бензол, карбонильная и карбоксильная группы и др. sp линейная Ацетилен и его гомологи, нитрил, кумулированные углеводороды и др.
Лекция № 3 Изомерия Изомеры – это вещества, имеющие одинаковый состав и молекулярную массу, но разные физические и химические свойства. Различия в свойствах изомеров обусловлены различиями в их химическом или пространственном строении. Под химическим строением понимают природу и последовательность связей между атомами в молекуле. Изомеры, молекулы которых отличаются по химическому строению, называют структурными изомерами. 1. Структурная изомерия Структурные изомеры могут отличаться:
Геометрическая изомерия Геометрическая изомерия характерна для соединений, в которых отсутствует вращение вокруг связи. Это могут быть соединения с двойной связью или циклические. Важное следствие жесткости двойной связи (отсутствия вращения вокруг нее) - существование геометрических изомеров. Самые распространенные из них - это цис-,транс-изомеры соединений этиленового ряда, содержащих у ненасыщенных атомов неодинаковые заместители. Простейшим примером могут служить изомеры бутена-2.
Геометрические изомеры имеют одинаковое химическое строение, различаются по пространственному расположению атомов, т.е. конфигурациями. Это различие и создает разницу в физических и химических свойствах. Геометрические изомеры, в отличие от конформеров, могут быть выделены в чистом виде и существуют как индивидуальные устойчивые вещества. Для их взаимного превращения необходима энергия порядка 125-170 кДж/моль, которую можно сообщить нагреванием или облучением. В простейших случаях номенклатура геометрических изомеров не представляет затруднений: цис- формами называют геометрические изомеры, у которых одинаковые заместители лежат по одну сторону от плоскости π-связи, транс- изомеры имеют одинаковые заместители на разных сторонах от плоскости π-связи. В более сложных случаях применяется Z,E-номенклатура. Ее главный принцип: для обозначения конфигурации указывают цис- (Z, от немецкого Zusammen - вместе) или транс- (Е, от немецкого Entgegen - напротив) расположение старших заместителей при двойной связи. В Z,E-системе старшими считаются заместители с большим атомным номером. Если атомы, непосредственно связанные с ненасыщенными углеродами, одинаковы, то переходят ко "второму слою", в случае необходимости - к "третьему слою" и т.д (номенклатура Канна, Ингольда, Прелога – КИП). Рассмотрим применение правил Z,E-номенклатуры на двух примерах.
Начнем с формулы I, где все решается атомами "первого слоя". Расставив их атомные номера, получим, что старшие заместители каждой пары (бром в верхней части формулы и азот в нижней) находятся в транс -положении, отсюда следует стереохимические обозначение Е:
Е-1-бром-1-хлор-2-нитроэтен Для определения стереохимического обозначения структуры II необходимо искать различие в "высших слоях". По первому слою группы СН3, С2Н5, С3Н7 не отличаются. Во втором слое у группы СН3 сумма атомных номеров равна трем (три атома водорода), у групп С2Н5 и С3Н7 - по 8. Значит, группа СН3 не рассматривается - она младше двух других. Таким образом, старшие группы - это С2Н5 и С3Н7, они находятся в цис -положении; стереохимическое обозначение Z.
Z-3-метилгептен-3 Если бы понадобилось определить, какая группа старше - С2Н5 или С3Н7, пришлось бы перейти к атомам "третьего слоя", сумма атомных номеров в этом слое для обеих групп оказались бы соответственно равными 3 и 8, т.е. С3Н7 старше, чем С2Н5. В более сложных случаях определения старшинства надо учитывать дополнительные условия, как-то: атом, связанный двойной связью, считается дважды, связанный тройной - трижды; из числа изотопов старше более тяжелый (дейтерий старше водорода) и некоторые другие. Отметим, что обозначения Z не является синонимами цис- обозначений, как и обозначения Е не всегда соответствуют расположению транс-, например:
Для 1,2-диметилциклопропана геометрические изомеры можно представить следующим образом:
цис- 1,2-диметилциклопропан транс- 1,2-диметилциклопропан Проекционные формулы Для условного изображения асимметрического атома на плоскости пользуются проекционными формулами Э.Фишера. Их получают, проецируя на плоскость атомы, с которыми связан асимметрический атом. При этом сам асимметрический атом, как правило, опускают, сохраняя лишь перекрещивающиеся линии и символы заместителей. Чтобы помнить о пространственном расположении заместителей, часто сохраняют в проекционных формулах прерывистую вертикальную линию (верхний и нижний заместитель удалены за плоскость чертежа), однако часто этого не делают. Ниже приведены различные способы записи проекционной формулы, отвечающей левой модели на предыдущем рисунке:
Приведем несколько примеров проекционных формул:
При названиях веществ приведены их знаки вращения. Это значит, например, что левовращающий антипод бутанола-2 имеет пространственную конфигурацию, выражаемую именно приведенной выше формулой, а ее зеркальное изображение отвечает правовращающему бутанолу-2. Определение конфигурации оптических антиподов проводится экспериментально. Смесь, состоящая из равного количества обоих антиподов, называется рацематом. Она оптически неактивна, имеет отличные значения физических констант. В принципе, каждый оптический антипод может быть изображен двенадцатью (!) различными проекционными формулами - в зависимости от того, как расположена модель при построении проекции, с какой стороны мы смотрим на нее. Чтобы стандартизировать проекционные формулы, введены определенные правила их написания. Так, главную функцию, если она находится в конце цепи, принято ставить наверху, главную цепь изображать вертикально. Правила преобразования проекционных формул: 1. Формулы можно вращать в плоскости чертежа на 180о, не меняя их стереохимического смысла:
2. Две (или любое четное число) перестановки заместителей у одного асимметрического атома не меняют стереохимического смысла формулы:
3. Одна (или любое нечетное число) перестановок заместителей у асимметрического центра приводит к формуле оптического антипода:
4. Поворот в плоскости чертежа на 90о превращает формулу в антипод, если только при этом одновременно не изменить условие расположения заместителей относительно плоскости чертежа, т.е. не считать, что теперь боковые заместители находятся за плоскостью чертежа, а верхний и нижний - перед ней. Если пользоваться формулой с пунктиром, то изменившаяся ориентация пунктира прямо напомнит об этом:
5. Вместо перестановок проекционные формулы можно преобразовывать путем вращения любых трех заместителей по часовой стрелке или против нее; четвертый заместитель при этом положения не меняет (такая операция эквивалентна двум перестановкам):
6. Проекционные формулы нельзя выводить из плоскости чертежа. Другие типы оптически активных веществ В этом разделе перечислены некоторые другие классы органических соединений, также обладающих оптической активностью (т.е. существующие в виде пар оптических антиподов). Атом углерода не обладает монополией на создание хиральных центров в молекулах органических соединений. Центром хиральности могут быть также атомы кремния, олова, четырехковалентного азота в четвертичных аммониевых солях и окисях третичных аминов:
В этих соединениях центр асимметрии имеет тетраэдрическую конфигурацию, как и асимметрический атом углерода. Существуют, однако, и соединения с иной пространственной структурой хирального центра. Пирамидальную конфигурацию имеют хиральные центры, образованные атомами трехвалентного азота, фосфора, мышьяка, сурьмы, серы. В принципе, центр асимметрии можно считать тетраэдрическим, если в качестве четвертого заместителя принять неподеленную электронную пару гетероатома:
Оптическая активность может возникать и без хирального центра, за счет хиральности структуры всей молекулы в целом (молекулярная хиральность или молекулярная асимметрия). Наиболее характерными примерами являются наличие хиральной оси либо хиральной плоскости. Хиральная ось возникает, например, в алленах, содержащих различные заместители при sp2 -гибридных углеродных атомах. Легко видеть, что приведенные ниже соединения являются несовместимыми зеркальными изображениями, а значит оптическими антиподами:
Другой класс соединений, имеющих хиральную ось - оптически активные бифенилы, которые имеют в орто -положениях объемистые заместители, затрудняющие свободное вращение вокруг С-С связи, соединяющей бензольные ядра:
Хиральная плоскость характеризуется тем, что у нее можно различить "верх" и "низ", а также "правую" и "левую" стороны. Примером соединений с хиральной плоскостью могут служить оптически активный транс- циклооктен и оптически активное производное ферроцена:
Диастереомерия Соединения с несколькими асимметрическими атомами обладают важными особенностями, отличающими их от рассмотренных ранее более простых оптически активных веществ с одним центром асимметрии. Допустим, что в молекуле некоего вещества имеются два асимметрических атома; обозначим их условно А и Б. Легко видеть, что возможны молекулы со следующими комбинациями (N=22=4):
Молекулы 1 и 2 представляют собой пару оптических антиподов; то же самое относится и к паре молекул 3 и 4. Если же сравнивать друг с другом молекулы из разных пар антиподов - 1 и 3, 1 и 4, 2 и 3, 2 и 4, то мы увидим, что перечисленные пары не являются оптическими антиподами: конфигурация одного асимметрического атома у них совпадает, конфигурация другого - не совпадает. Это пары диастереомеров, т.е. пространственных изомеров, не составляющих друг с другом оптических антиподов. Диастереомеры отличаются друг от друга не только оптическим вращением, но и всеми другими физическими константами: у них разные температуры плавления и кипения, разные растворимости и др. Различия в свойствах диастереомеров зачастую ничуть не меньше, чем различия в свойствах между структурными изомерами. Примером соединения рассматриваемого типа может случить хлоряблочная кислота(2 различных хиральных центра):
Ее стереоизомерные формы имеют следующие проекционные формулы:
Названия эритро - и трео - происходят от названий углеводов эритрозы и треозы. Эти названия употребляют для указания взаимного положения заместителей у соединений с двумя асимметрическими атомами: эритро -изомерами называют те, у которых два одинаковых боковых заместителя стоят в стандартной проекционной формуле на одной стороне (справа или слева, два конца буквы Э); трео -изомеры имеют одинаковые боковые заместители на разных сторонах проекционной формулы. Два эритро- изомера представляют собой пару оптических антиподов, при их смешении образуется рацемат. Парой оптических изомеров являются и трео- формы; они тоже дают при смешении рацемат, отличающийся по свойствам от рацемата эритро- формы. Таким образом, всего существуют четыре оптически активных изомера хлоряблочной кислоты и два рацемата. Число стереоизомеров может уменьшаться из-за частичной симметрии, появляющейся в некоторых структурах. Примером может служить винная кислота, у которой число индивидуальных стереоизомеров сокращается до трех. Их проекционные формулы:
Формула I идентична с формулой Iа, так как превращается в нее при повороте на 180о в плоскости чертежа и, следовательно, не изображает нового стереоизомера. Это оптически неактивная модификация называется мезо-форма. Мезо-формы имеются у всех оптически активных веществ с несколькими одинаковыми (т.е. связанными с одинаковыми заместителями) асимметрическими центрами. Проекционные формулы мезо-форм всегда можно узнать по тому, что их можно разделить горизонтальной линией на две половины, которые по записи на бумаге формально идентичны, в действительности же зеркальны (плоскость внутренней симметрии):
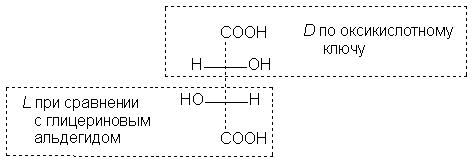
Формулы II и III изображают оптические антиподы винной кислоты; при их смешении образуется оптически неактивный рацемат - виноградная кислота. Номенклатура оптических изомеров Самая простая, наиболее старая, однако и ныне еще употребляемая система номенклатуры оптических антиподов, основана на сравнении проекционной формулы называемого антипода с проекционной формулой некоего стандартного вещества, выбранного в качестве "ключа". Так, для α -оксикислот и α -аминокислот ключом является верхняя часть их проекционной формулы (в стандартной записи):
Конфигурацию всех α -оксикислот, имеющих в стандартно написанной проекционной формуле Фишера гидроксильную группу слева, обозначают знаком L; если же гидроксил расположен в проекционной формуле справа - знаком D Ключом для обозначения конфигурации соединений служит глицериновый альдегид:
В молекулах сахаров обозначение D- или L- относится к конфигурации нижнего асимметрического центра. Система D-, L- обозначений имеет существенные недостатки: во-первых, обозначение D- или L- указывает конфигурацию только одного асимметрического атома, во-вторых, для некоторых соединений получаются разные обозначения, в зависимости от того, взят ли в качестве ключа глицериновый альдегид или оксикислотный ключ, например:
Эти недостатки системы ключей ограничивают ее применение в настоящее время тремя классами оптически активных веществ: сахарами, аминокислотами и оксикислотами. На общее же применение рассчитана R,S-система Кана, Ингольда и Прелога. Для определения R- или S-конфигурации оптического антипода необходимо расположить тетраэдр заместителей вокруг асимметрического углеродного атома таким образом, чтобы младший заместитель (обычно это водород) имел направление "от наблюдателя". Если движение при переходе по кругу трех остальных заместителей от старшего к среднему по старшинству и затем к самому младшему происходит против часовой стрелки - это S -изомер (ассоциируется с таким же движением руки при написании буквы S), если по часовой стрелке - это R- изомер (ассоциируется с движением руки при написании буквы R). Для определения старшинства заместителей у асимметрического атома используются правила подсчета атомных номеров, уже рассмотренные нами в связи с Z,E-номенклатурой геометрических изомеров. Для выбора R,S-обозначений по проекционной формуле необходимо путем четного числа перестановок (не изменяющих, как мы знаем, стереохимического смысла формулы) расположить заместители так, чтобы младший из них (обычно водород) оказался внизу проекционной формулы. Тогда старшинство остальных трех заместителей, падающее по часовой стрелке, соответствует обозначению R, против часовой стрелки - обозначению S:
Лекция № 4 Кислоты и основания Льюиса Дж. Льюисом была предложена более общая теория кислот и оснований. Основания Льюиса – это доноры пары электронов (спирты, алкоголят-анионы, простые эфиры, амины и т.д.) Кислоты Льюиса – это акцепторы пары электронов, т.е. соединения, имеющие вакантную орбиталь (ион водорода и катионы металлов: H+, Ag+, Na+, Fe2+; галогениды элементов второго и третьего периодов BF3, AlCl3, FeCl3, ZnCl2; галогены; соединения олова и серы: SnCl4, SO3). Таким образом, основания Бренстеда и Льюиса – это одни и те же частицы. Однако основность по Бренстеду есть способность присоединять только протон, в то время как основность по Льюису – понятие более широкое и означает способность к взаимодействию с любой частицей, имеющей низколежащую свободную орбиталь. Кислотно-основное взаимодействие по Льюису есть доноро-акцепторное взаимодействие и любую гетеролитическую реакцию можно представить как взаимодействие кислоты и основания Льюиса:
Единой шкалы для сравнения силы кислот и оснований Льюиса не существует, так как их относительная сила будет зависеть от того, какое вещество взято за стандарт (для кислот и оснований Бренстеда таким стандартом является вода). Для оценки легкости протекания кислотно-основного взаимодействия по Льюису Р. Пирсоном была предложена качественная теория “жестких” и “мягких” кислот и оснований. Жесткие основания обладают высокой электроотрицательностью и низкой поляризуемостью. Они трудно окисляются. Их высшие занятые молекулярные орбитали (ВЗМО) имеют низкую энергию. Мягкие основания имеют низкую электроотрицательность и высокую поляризуемость. Они легко окисляются. Их высшие занятые молекулярные орбитали (ВЗМО) имеют высокую энергию. Жесткие кислоты имеют высокую электроотрицательность и низкую поляризуемость. Они трудно восстанавливаются. Их низшие свободные молекулярные орбитали (НСМО) имеют низкую энергию. Мягкие кислоты обладают низкой электроотрицательностью и высокой поляризуемостью. Они легко восстанавливаются. Их низшие свободные молекулярные орбитали (НСМО) имеют высокую энергию. Самая жесткая кислота - Н+, самая мягкая – СН3Hg+. Наиболее жесткие основания – F- и OH-, наиболее мягкие – I- и Н-. Таблица 5. Жесткие и мягкие кислоты и основания.
Принцип жестких и мягких кислот и оснований Пирсона (принцип ЖМКО): Жесткие кислоты преимущественно взаимодействуют с жесткими основаниями, а мягкие кислоты – с мягкими основаниями. Это выражается в больших скоростях реакций и в образовании более устойчивых соединений, так как взаимодействие между близкими по энергии орбиталями эффективнее, чем взаимодействие между орбиталями, значительно различающимися по энергии. Принцип ЖМКО используют для определения преимущественного направления конкурирующих процессов (реакции элиминирования и нуклеофильного замещения, реакции с участием амбидентных нуклеофилов); для направленного создания детоксикантов и лекарственных препаратов. Лекция № 5 Карбкатионы представляют собой положительно заряженные частицы с тремя заместителями при центральном атоме углерода, имеющем одну вакантную несвязывающую орбиталь. Карбанионы – отрицательно заряженные частицы с тремя заместителями при центральном атоме углерода, имеющем несвязывающую орбиталь с парой электронов. Выделение в механизме реакций скорость-определяющей стадии имеет принципиальное значение, поскольку все факторы, влияющие на эту стадию, аналогичным образом влияют на скорость реакции в целом. При оценке влияния различных факторов на скорость реакции оценивают их влияние на энергии исходного и переходного состояний скорость-определяющей стадии. Факторы, стабилизирующие переходное состояние в большей степени, чем исходное, уменьшают энергию активации и увеличивают скорость реакции, и наоборот. Переходные состояния имеют практически нулевое время жизни, поэтому их структуру нельзя определить экспериментально. Она может быть оценена теоретически, путем сравнения их с реальными частицами, близкими к ним по энергии. Такими частицами могут быть интермедиаты, которые можно рассматривать как модели переходных состояний. Если хотя бы одна из конкурирующих реакций обратима, то соотношение продуктов может быть иным. Если остановить реакцию задолго до достижения равновесия, то реакция будет подчиняться кинетическому контролю и основным ее продуктом будет вещество С. Если же довести процесс до состояния равновесия, то в реакционной смеси будет преобладать термодинамически более устойчивый продукт В. В этом случае реакция подчиняется термодинамическому контролю. На рисунке приведен случай несовпадения кинетического и термодинамического контроля продуктов реакции. Возможен случай, когда термодинамически устойчивый продукт образуется с большей скоростью, т. е. является и кинетически, и термодинамически контролируемым. УГЛЕВОДОРОДЫ Лекция 6 АЛКАНЫ Алканы – это ациклические углеводороды, содержащие только простые связи С-С. Общая формула – СnH2n+2. Методы получения
cat: Ni, Pt, Pd
2RCOONa + 2H2O →R-R + 2CO2 + 2NaOH + H2 Химические свойства Алканы содержат σ -связи С-Н и С-С, для которых характерны высокая прочность (ЕС-С=347 кДж/моль; ЕС-Н=415 кДж/моль), малая полярность (µС-С = 0D; µС-Н = 0,4D), низкая поляризуемость. Алканы обладают низкой реакционной способностью. Их превращения осуществляются в жестких условиях. При этом почти всегда происходит гомолитический разрыв связей, который требует существенно меньших затрат энергии. Основной тип реакций алканов – реакции радикального замещения: 1. Галогенирование Галогенирования алканов протекает при УФ-облучении или высокой температуре по схеме: RH + Hal2 →RHal + HHal Hal=Cl, Br. Реакционная способность галогенов по отношению к алканам уменьшается в ряду: F2 >Cl2 >Br2 >I2 Фторирование в обычных условиях приводит к полному разрушению органической молекулы. Иод с алканами не взаимодействует. Реакции галогенирования протекают по цепному радикальному механизму, который включает следующие стадии: инициирование цепи, рост цепи,обрыв цепи. Стадией, определяющей скорость реакции, является стадия отщепления водорода атомом галогена. При хлорировании углеводородов, как правило, образуется смесь монохлорпроизводных, т.е. реакции протекают неселективно. Зная относительную реакционную способность С-Н связей (перв -С-Н < втор -C-Н < трет- С-Н) и количество С-Н связей определенного сорта, можно рассчитать состав смеси. Указанный ряд соответствует ряду относительной стабильности алкильных радикалов:
Бромирование алканов, в отличие от хлорирования, протекают региоселективно, в первую очередь образуются третичные бромпроизводные. 2. Сульфохлорирование Под действием смеси оксида серы (IV) и хлора при УФ-облучении из алканов образуются алкилсульфохлориды: RH + SO2 + Cl2 → RSO2Cl + HCl Реакция протекает по свободнорадикальному механизму. При обработке алкилсульфохлоридов щелочами образуются акилсульфонаты, которые используются как поверхностно-активные вещества (ПАВ): RSO2Cl + 2NaOH → RSO3-Na+ + NaCl + H2O Алкилсульфонат натрия 3. Нитрование При нагревании алканов с разбавленной азотной кислотой в жидкой фазе (М.И.Коновалов) происходит образование нитросоединений:
Замещение происходит преимущественно у наиболее реакционноспособного третичного атома углерода. Например:
В промышленности нитрование алканов проводят в газовой фазе при 150-4500С парами азотной кислоты. Газофазное нитрование протекает неселективно с образованием наряду с продуктами замещения водорода продуктов продуктов расщепления С-С связей:
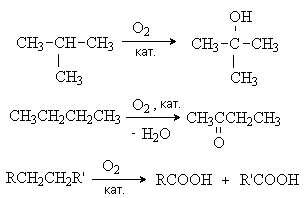
4. Окисление При комнатной температуре даже такие сильные окислители, как KMnO4 и K2Cr2O7, не действуют на алканы. При высоких температурах в присутствии избытка кислорода алканы сгорают с образованием углекислого газа и воды: 2CnH2n+2 + (3n + 1) O2 → 2n CO2 + (2n + 2) H2O В более мягких условиях (температура не выше 2000С) в присутствие катализаторов (соединений марганца) происходит неполное окисление алканов кислородом с образованием спиртов, кетонов, карбоновых кислот. В реакции принимают участие наиболее реакционноспособные третичные или вторичные С-Н связи:
Реакции используются в промышленности для синтеза ценных кислородсодержащих соединений и их смесей. Лекция 7 НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ А л к е н ы Алкены – это углеводороды состава СnH2n, содержащие в молекуле одну связь С=С. Методы получения
Химические свойства Химические свойства алкенов определяются наличием связи С=С. π -Связь обладает меньш
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-09-18; просмотров: 2047; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.189.171.188 (0.019 с.) |