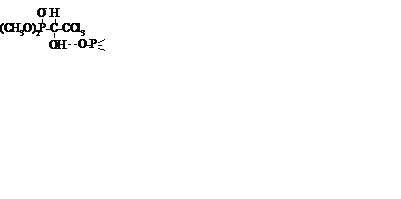Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Методы синтеза эфиров a –оксифосфоновых кислотСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
К большому классу фосфорорганических соединений, относятся моно- (1.37) и дизамещенные (1.38) эфиры фосфористых кислот:
(1.37) (1.38)
Соединения (1.38) имеют огромное значение в химии фосфорорганических соединений. Фосфиты (1.38) легко депротонируются с образованием анионов, которые проявляют высокую нуклеофильность и могут алкилироваться как амбидентные системы по фосфору или по кислороду. Направление реакции алкилирования определяется соответствием мягко-жестких характеристик нуклеофильных фрагментов фосфит-аниона и электрофила. В обычных условиях чаще всего алкилируется атом фосфора и образуются эфиры a–оксифосфоновых кислот (фосфонаты) [70–74]. Основным методом синтеза эфиров a–оксифосфоновых кислот, впервые предложенным В.С. Абрамовым в 1950 году, является непосредственное действие альдегидов и кетонов на соли диалкилфосфористых кислот. Описанные до этого методики синтеза таких соединений отличались, как правило, небольшими выходами целевых продуктов реакции [75,76]. Предложенный В.С. Абрамовым и в дальнейшем разработанный новый метод получения a–оксиалкилфосфоновых кислот сделал их доступными соединениями, и в настоящее время он широко используется в химии фосфорорганических соединений. На большом экспериментальном материале было показано, что присоединение диалкилфосфористых кислот и других типов неполных эфиров кислот фосфора является характерной реакцией для альдегидов и кетонов [78].
Реакция является общей для окисей фосфинов и эфиров кислот фосфора, содержащих группу Р(О)Н. Как правило, реакция присоединения диалкил-фосфитов по карбонильной группе осуществляется в присутствии каталитических количеств сильного основания, преимущественно алкоголятов щелочных металлов. Реакции присоединения неполных эфиров кислот (1.38) фосфора по карбонильной группе (1.39) в присутствии алкоголятов щелочных металлов протекают по ионному механизму в соответствии со схемой [75]:
Изображая схему в общей (синтонной) форме и в символах концепции жестких и мягких кислот и оснований (ЖМКО) Пирсона, реакцию рассматривают как цепную, при этом алкоголят – анион RO- является не катализатором, а инициатором, «погибающим» в стадии инициирования. Цепь может продолжаться до исчерпания реагентов или исчезновения одного из интермедиатов в побочной реакции (обрыв цепи).
Наряду с алкоголятами, в качестве инициаторов реакции, могут выступать мягкие нуклеофильные реагенты, средние фосфиты и вторичные амины. Во многих, а возможно и во всех случаях, реакции протекают и в отсутствие катализаторов, при длительном выдерживании реакционной смеси при комнатной температуре или нагревании. Иногда реакции сопровождаются экзотермическим эффектом. В случае присоединения оксидов вторичных фосфинов, отмечен случай автокатализа. Полнота течения реакции присоединения диалкилфосфитов зависит от радикалов у карбонильной группы: если радикалы усиливают эффект поляризации карбонильной группы, реакция проходит энергично и полно, если же радикалы уменьшают эффект поляризации, то реакция проходит умеренно, а в некоторых случаях не идет вообще. Было показано, что присоединение неполных эфиров кислот фосфора значительно облегчается в случае a – галоидкарбонильных соединений и других производных альдегидов и кетонов, содержащих электроотрицательные группы. Особенно энергично протекают реакции с ди-, три- и полихлорзамещенными карбонильными соединениями [76]. В отсутствие катализатора может успешно происходить и реакция диметилфосфита с хлоралем [77 – 80]. Наиболее важным вторичным процессом, дополняющим реакцию Абрамова, является фосфонат – фосфатная перегруппировка:
По-видимому, она заключается во внутримолекулярном нуклеофильном замещении у атома фосфора под действием алкоксианиона. В соответствии с этим скорость перегруппировки зависит от целого ряда факторов, среди которых наиболее важны такие, как наличие дефицита электронной плотности на атоме фосфора, нуклеофильность атома кислорода гидроксильной группы, прочность связи фосфор – углерод, пространственные и другие эффекты. Полагают, что в условиях щелочного катализа или в отсутствие его при более высоких температурах распад на исходные реагенты и фосфонат – фосфатная перегруппировка являются для эфиров a –оксифосфоновых кислот конкурирующими реакциями, конечный результат которых зависит от целого ряда факторов. Известно, что реакция Абрамова может сопровождаться побочными процессами, например:
Несмотря на то, что за последние десятилетия реакция присоединения неполных эфиров кислот фосфора к карбонильным соединениям получила широкую известность и систематически изучается, сведения о ее механизме противоречивы [81,82]. В.С. Абрамовым с сотрудниками было сделано предположение, что в тех случаях, когда присоединение диалкилфосфористых кислот к карбонильным соединениям протекает в отсутствии катализатора, кислота реагирует в енольной форме (1.40), вследствие достаточно высокой скорости взаимодействия минимального количества енольной (фосфитной) формы (1.40) с реакционноспособными карбонильными соединениями.
Электрофильный атом углерода карбонильной группы взаимодействует с нуклеофильным атомом фосфора, несущим необобщенную пару электронов с образованием эфиров a-оксиалкилфосфоновых кислот (1.41):
Интересно отметить, что ряд диалкилфосфитов, содержащих атомы хлора в b-положении алкильной группы, как например, ди(трет-b,b,b –трихлорбутил)фосфористая кислота реагирует с простыми карбонильными соединениями без катализатора [78-80]. В данном случае, как полагают авторы, электроноакцепторные атомы хлора в радикалах диалкилфосфитов оказывают влияние на таутомерное равновесие, сдвигая его в сторону образования фосфитной формы. Возможно, что последняя присутствует в этих эфирах в необычно высокой концентрации, поскольку расположение весьма объемных заместителей в вершинах тетраэдрической конфигурации молекулы диалкилфосфита, приводит к тому, что они оказываются пространственно достаточно сближенными для осуществления преимущественного взаимодействия друг с другом и в то же время достаточно удаленными от центрального атома фосфора, чтобы существенно дезактивировать его посредством индуктивного оттягивания электронов [81]. Однако, считая, что диалкилфосфористые кислоты взаимодействуют в отсутствии катализаторов в своей енольной форме (1.40), авторы высказывают некоторые возражения. Предполагается, что реакционная способность трехвалентного фосфора является максимальной именно в тетраэдрической конфигурации с неподеленной парой электронов на sp3–гибридной орбитали и с геометрией по существу той же самой, что и в переходном состоянии. Показано, что триалкилфосфиты, в отличие от диалкилфосфитов, не образуют устойчивых продуктов присоединения с простыми карбонильными соединениями в отсутствии основного катализатора. Возможно, что диалкилфосфиты присоединяются обратимо и необходим перенос протона, чтобы стабилизировать аддукт реакции. Хотя присоединение диалкилфосфитов по карбонильной группе и может быть обратимым, все же в обычных условиях равновесие сдвинуто в сторону образования оксифосфоната. В.С. Абрамовым было выдвинуто предположение [82], что реакция протекает с диалкил-анионом (1.421), так как присутствие сколько-нибудь значительного количества енольной формы диалкилфосфита не подтвердилось данными ИК-спектроскопии. При этом обе таутомерные формы неполных фосфитов кислот фосфора образуют общий ион, который далее вступает в реакцию с электрофильным атомом карбонильной группы
В работах [83,84] изучению влияния различных растворителей на кинетику реакции конденсации диметилфосфита (1.43) с хлоралем (1.44) обсуждаются возможные механизмы реакции в различных средах без добавления катализатора:
Сравнение величин констант скорости реакции диметилфосфита с хлоралем в различных неполярных (CCl4, гексан, бензол) и диполярных апротонных растворителях (диоксан, тетрагидрофуран, ацетонитрил, анизол, диметилсульфоксид) показало, что с увеличением полярности растворителя скорость реакции уменьшается. Как объясняют авторы, в этом случае действуют два фактора: увеличение полярности растворителя способствует сдвигу таутомерного равновесия в сторону реакционно - способной таутомерной формы с трехвалентным фосфором и одновременно увеличивает специфическую сольватацию за счет упрочения водородных связей этой формы с растворителем. Конкуренцию этих двух факторов, действующих в противоположных направлениях, качественно учесть трудно. Эти данные не противоречат тому факту, что в реакциях присоединения триалкилфосфитов по карбонильной группе наблюдается обратное явление, т.е. увеличение скорости реакции с ростом полярности среды. В отличие от диалкилфосфитов, триалкилфосфиты не образуют водородные связи с растворителем и в этом случае, действует фактор ускоряющего влияния полярности среды. Было сделано предположение, что в различных средах могут реализовываться два возможных механизма присоединения диалкилфосфитов к карбонильным соединениям: через диалкилфосфит − анион (1.42) или через трехвалентную форму (1.40) диалкилфосфита. Так, в среде инертных и основных недиссоциирующих растворителей реализуется механизм через трехвалентную фосфитную форму, при этом такие полярные растворители, как ацетонитрил, диоксан, тетрагидрофуран, анизол уменьшают скорость взаимодействия. Если бы реакция во всех средах протекала через диалкилфосфит − анион, то полярные растворители должны были бы резко увеличивать скорость процесса. Однако такое увеличение наблюдалось лишь в растворе диметилсульфоксида. Это позволило сделать предположение, что в этом растворителе реакция идет через диалкилфосфит − анион. Механизм процесса определяется условиями, в частности соотношением реагентов и природой растворителя. Анализ кинетических и спектральных данных, полученных при взаимодействии хлораля (1.44) и диметилфосфита (1.43) в отсутствии катализатора, как без растворителя, так и в апротонных растворителях, позволил предложить новую схему протекания реакции, которая не противоречит ранее описанным наблюдениям. Эта схема учитывает роль диметилфосфита как акцептора протонов, а также роль других акцепторов: триэтиламина, диметилсульфоксида, диоксана, тетрагидрофурана и т.д. В соответствии с этой схемой суммарный порядок реакции с участием различных акцепторов может меняться от второго до третьего при изменении растворителя или концентрации диметилфосфита. Роль акцепторов протонов заключается в освобождении электронной пары трехвалентного фосфора в промежуточном соединении типа «полуацеталя» (1.45), который затем за счет внутримолекулярного алкилирования – синхронного смещения двух электронных пар от нуклеофильного фосфора с одной стороны и от углерода к кислороду у атома фосфора с другой стороны превращается в конечный продукт (1.46). Эта стадия аналогична внутримолекулярной перегруппировке Арбузова:
1.47 Наиболее существенной частью этой схемы является роль в реакции «полуацеталя» (1.45), который и наблюдается в ИК-спектрах. Образование «полуацеталя» может проходить при непосредственном взаимодействии хлораля и диметилфосфита с четырехкоординационным атомом фосфора через циклический переходной комплекс (1.48), наподобие 1,3 − диполярного присоединения к карбонильной группе:
Такую структуру нужно воспринимать не как анион трехвалентной формы, а как одну из мезомерных форм структуры четырехкоординационного фосфита. Образование малополярного циклического комплекса (1.45) легко объясняет такие факторы, как замедление реакции с увеличением полярности растворителя и его кислотности. Необходимо отметить, что в избытке хлораля кинетические закономерности осложняются за счет образования полуацеталя хлорофоса (1.49), что ведет к существенному торможению процесса по мере его накопления.
1.49 Однако при повышении температуры до 50ºС, реакция практически доходит до конца, за счет диссоциации полуацеталя (1.49). Найдена зависимость реакционной способности гидрофосфорильной группы от ее ориентации. Так, в реакции с 4-бромбензальдегидом и трифторметил–4–толилкетоном 1,3-бутиленфосфит с аксиально ориентиро-ванным фосфорилом существенно более активен (k×102 соответственно 0,20 и 3,2), чем 1,3–бутиленфосфит с экваториально ориентированным фосфорилом (k×102 соответственно 0,11 и 2,3). Изучив с помощью ЯМР 13С и ЯМР 31Р стереохимию карбоциклических гидроксифосфоновых соединений, авторы пришли к выводу, что для ряда циклогексильных соединений величины вицинальной связи между С–3 и С–5 позиций кольца указывают на преобладание конформеров с экваториальным фосфорилом и аксиальной ОН–группой. Установлено также экваториальное расположение СН3-группы для о–, м–, п – метилциклогексильных производных. Наиболее подробно реакция Абрамова изучена на примере диалкилфосфитов, неполных фосфонитов и фосфонистых кислот. В последние годы стали использоваться и другие классы гидрофосфорильных соединений [84]. На примере конденсации диалкилфосфористых кислот с различными непредельными альдегидами и кетонами А.Н Пудовик с сотрудниками установили, что в отличие от a–непредельных кетонов этиленового ряда, присоединение диалкилфосфитов в присутствии алкоголятов натрия происходит не по кратным связям, а по карбонильной группе, и полученные продукты являлись непредельными эфирами a–оксифосфоновых кислот. Присоединение наблюдалось в тех случаях, когда электрофильные свойства карбонильной группы усиливались электроотрицательными заместителями [85-87]. Известны реакции присоединения диалкилфосфитов к гетероциклическим кетонам с характером различного замещения в гетероцикле.
|
||||
|
|
Последнее изменение этой страницы: 2016-08-10; просмотров: 474; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.146.176.191 (0.009 с.) |