
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Розчини високомолекулярних сполукСодержание книги
Поиск на нашем сайте υ1 = υ2; то k1С1С2 = k2С1’C2’. При перенесенні константних величин вліво, а змінних – вправо, одержуємо рівняння:
Частка від ділення сталої величини на сталу величину буде сталою:
Наведені рівняння є рівняннями рівноваги оборотної хімічної реакції. Стала k називається константою рівноваги. Отже, константою рівноваги називають відношення добутку концентрацій продуктів реакції до добутку концентрацій вихідних реагуючих речовин. Спряжені реакції характерні тим, що одна реакція відбувається тільки в присутності іншої реакції. Для цих реакцій характерна наступна схема: А+В → М (а); А+С → N (б). Ці дві реакції зв’язані між собою спряжено – перебіг другої реакції індукується першою. Спільну речовину для обох (А) називають актором, речовину С – акцептором, речовину В – індуктором. Перша реакція проходить самочинно і називається первинною, індукована друга реакція – вторинною. Приклад спряженої реакції – окиснення ферум (ІІ) сульфату і гідроген йодиду пероксидом. Гідроген йодид пероксидом не окиснюється, але при окисненні ферум (ІІ) сульфату він окиснюється разом з ним. Тому, перша реакція (І) відбувається самостійно, тоді як друга (ІІ) проходить лиш у присутності (І): Н2О2+2FeSO4+H2SO4 → Fe2(SO4)3+2H2O; (I) H2O2+2HJ → 2H2O+J2. (II) У цих реакціях Н2О2 актор; FeSO4 – індукує (ІІ) реакцію, індуктор; HJ – акцептор. У біологічному світі прикладом спряжених реакційних систем може бути дихальний ланцюг, зв’язаний з окиснювальним фосфорилюванням. Особливе значення мають ланцюгові реакції . Вони є складними хімічними або ядерними процесами, при яких поява проміжної активної частинки (у хімічних реакціях – збуджені молекули, радикали чи атоми, в ядерних –нейтрон) викликає велику кількість (ланцюг) перетворень вихідних молекул або ядер. Їх поділяють на дві групи – розгалужені і нерозгалужені. Прикладом ланцюгової реакції нерозгалуженого типу може бути фотохімічна реакція між воднем і хлором. Молекула хлору, поглинаючи квант світла, розщеплюється на два радикали. Радикал хлору починає новий ланцюг хімічних перетворень. Довжина ланцюга одного з утвореного центру може досягати десятків і сотень тисяч. Обрив ланцюга відбувається на стінках реакційної посудини, які асимілюють енергію, що виникає при рекомбінації, і стабілізують молекулу.
Cl2+hγ → 2Cl· зародження ланцюга Cl·+H2 → HCl+H· H·+Cl2 → HCl+Cl·
} розвиток ланцюга 2Cl·+стінка → Сl2 Cl·+Cl·+M → Cl2+M обрив ланцюга, де: М – атом або молекула, яка поглинає енергію; hγ – квант світла (h – стала Планка, γ – частота випромінювання). Якщо замість кожного активного зниклого центру в середньому утворюється більше ніж один новий центр, то такі реакції називають розгалуженими. Приклад – вибух гримучого газу (суміш Н2 і О2 2:1):
§5. Хімічні реакції у біологічних системах. Процеси в біологічному світі належать до відкритих систем, у яких відбувається постійний обмін речовин і енергії із зовнішнім середовищем. Обмін речовин у відкритих системах забезпечує безперервне надходження і видалення різних метаболітів. І, як наслідок, у живому організмі багато реакцій не досягають стану динамічної рівноваги, як це характерно для замкнутих систем, наприклад, in vitro, а відбувається безперервно, перебуваючи у стані стабільної переваги прямих реакцій. Спряжені, послідовні і паралельні хімічні реакції також відбуваються у відкритих системах, але особливістю таких систем є багатоступінчасті зміни, що відбуваються у вигляді циклів біохімічних реакцій, як, наприклад, цикл трикарбонових кислот, орнітиновий цикл біосинтезу сечовини та ін. У процесі обміну речовин відбуваються реакції лінійного і розгалуженого характеру. Слід враховувати, що, на відміну від постійного повторення однотипного процесу у ланцюгових реакціях, в біологічних процесах молекула може бути втягнута у декілька різних реакцій. Вибір шляху хімічного перетворення, яким піде кожна молекула, значною мірою визначається умовами обміну речовин в організмі. Основними критеріями життєвих процесів є специфічні відмінності між біологічними і хімічними відкритими системами, до яких у перших можуть бути віднесені здатності до самовідтворення, постійний обмін з навколишнім середовищем і наявність координації між собою. Розрізняють екзоергічні реакції, які відбуваються зі зменшенням внутрішньої енергії, та ендоергічні, що супроводжуються збільшенням внутрішньої енергії. У біоенергетичному відношенні у живих організмах має місце лише вільна енергія. При біохімічних процесах, як правило, вільна енергія, яка заключена у вихідних речовинах, повністю не використовується і певна її частина залишається у новостворених речовинах. Наприклад, енергія, що вивільнюється при окисненні різних органічних сполук, у більшій своїй частині зв’язується з деякими високоенергетичними сполуками, які беруть участь у багатьох біохімічних процесах, виконуючи роль резервної і транспортної форми енергії. У високоенергетичних сполуках енергія розподілена не рівномірно, а сконцентрована в окремих зв’язках молекули, так званих макроергічних. Макроергічними зв’язками багаті різні етери фосфатної кислоти:
поліфосфати і пірофосфати, енолфосфати, тіоетери та ін. При гідролізі макроергічних зв’язків вивільнюється значна кількість енергії (від 6 до 12 ккал/моль); так, наприклад, в одній найбільш важливій з біологічної точки зору сполуці, яка бере участь в усіх обмінах речовин, аденозинтрифосфатній кислоті (АТФ)
при розриві першого і другого макроергічних зв’язків виділяється приблизно 12 ккал/моль, у той час як при гідролізі першого зв’язку (3) утворюється лише 2,5 ккал/моль.
§6. Каталіз. Види каталізу. Каталіз (від грецьк. katalysis – руйнування) – зміна швидкості хімічної реакції під впливом каталізаторів. Під каталізом розуміють прискорення реакції (позитивний каталіз), однак в окремих випадках мається на увазі гальмування реакції (негативний каталіз). Каталізаторами можуть бути неорганічні та органічні сполуки у будь-якому агрегатному стані. Зміна швидкості хімічної реакції за участю каталізатора пов’язана зі зниженням енергії активації процесу порівнянно з енергією активації відповідних реакцій без каталізатора. Якщо зростання швидкості реакції викликається каталізатором, який утворюється у хімічному процесі, то реакція називається аутокаталітичною (наприклад, процеси аутооксидації та ін.), а саме явище – аутокаталізом. Різні каталітичні реакції прийнято розділяти на реакції гомогенного, гетерогенного і мікрогетерогенного каталізу. При гомогенному каталізі реагуючі речовини і каталізатор знаходяться в одній фазі – рідкій або газоподібній. Межі фаз між реагуючими речовинами і каталізатором не існує. Принцип дії каталізатора при гомогенному каталізі полягає у тому, що каталізатор, вступаючи в реакцію з реагентами, утворює нестійкі проміжні сполуки, які потім розпадаються з регенерацією каталізатора за схемою: А+В+K ⇄ (А–В–K)* → D+K. Швидкість цієї реакції υ = kIICАСВСК пропорційна концентрації каталізатора. Ця каталітична реакція може відбуватися у дві стадії:
А+K ⇄ АK, (1)
АK+В При цьому можливі два випадки. У першому швидкість розпаду комплексу на каталізатор і вихідний продукт значно більша, ніж швидкість другої стадії, у якій утворюється кінцевий продукт. Тому концентрація комплексів, які називаються при такому типі каталізу комплексами Арреніуса, мала. У другому випадку швидкість розпаду комплексу рівна швидкості другої стадії реакції. Концентрація проміжного комплексу значна і стаціонарна. Комплекси такого типу називають комплексами Вант-Гоффа. Другий випадок, як більш типовий, слід розглянути детальніше. Оскільки проміжна сполука АK є у рівновазі з вихідними речовинами, то швидкості прямої (υ1) і оборотної (υ2) реакцій (1) повинні бути рівними. Швидкості υ1 і υ2 можна виразити відповідними кінетичними рівняннями типу (5.12): k1CА(C’K–C’АК) = k2C’АК, де: C’K, C’АК – концентрація каталізатора, який не вступив у реакцію; C’А, C’АК – рівноважні концентрації речовини А і проміжної сполуки відповідно. Із вищенаведеного рівняння можна знайти концентрацію проміжної сполуки:
Сумарна швидкість всього процесу (υ) визначається швидкістю найповільнішої стадії, у даному випадку – другої. Тоді: υ = k3C’АКСВ. Якщо у це рівняння ввести концентрацію проміжної сполуки, то воно набуде такого вигляду:
Це рівняння вказує на існування двох крайніх випадків: 1) якщо k1CA <<< k2, то 2) якщо k1CA >>> k2, то υ = k3CBCK. В обох випадках швидкість реакції пропорційна концентрації каталізатора. Сумарний порядок рекції за вихідними речовинами різний і буде рівний двом або одиниці. Прикладом гомогененного каталізу є реакція термічного розкладу етаналю
Енергія активації кожної стадії менша, ніж енергія активації некаталітичної реакції. До гомогенного каталізу належить багато реакцій кислотно-основної взаємодії, реакції комплексоутворення і окиснення-відновлення, численні реакції гідрування та ін. При кислотному каталізі кислота віддає протони молекулі субстрату. Це супроводжується внутрімолекулярним перетворенням субстрату, пов’язаним зі зміною характеру і розташуванням зв’язків та відщепленням протону від другої ділянки молекули і приєднанням його до спряженої основи. Прикладом може бути реакція енолізації кетонів:
де А – кислота; А– – основа. У гомогенному каталізі можуть брати участь як каталізатори йони металів. Механізм каталітичної дії йонів металу, як правило, базується на поперемінному окисненні-відновленні: Cu+ ↔ Cu2+; Fe2+ ↔ Fe3+. Гетерогенний каталіз відбувається на межі двох фаз, тобто реакція відбувається на поверхні каталізатора. Каталізатор у цих реакціях є самостійною фазою системи. Між реагуючими речовинами і каталізатором існує поверхня поділу, наприклад тверда речовина в рідині, рідка речовина в газі і т.п. Каталіз, що здійснюється у колоїдних розчинах, емульсіях, зависях, також належить до гетерогенного. Як каталізатори часто використовують метали (Pt, Ni, Pd, Cu та ін.), їх оксиди (Al2O3, Fe2O3, Cr2O3, ZnO), деякі солі. У промисловості до каталізаторів нерідко додають промотори – речовини, які у невеликих кількостях (до 1%) підвищують активність каталізатора. Наприклад, при одержанні метану в присутності нікелю відбувається хімічна реакція: СО2+4Н2 Якщо в реакційну суміш ввести 0,5% церію, швидкість реакції зростає у 10 разів. Церій тут виконує роль промотора. Речовини, що знижують швидкість реакції, називаються інгібіторами. Вони використовуються для гальмування небажаних процесів: корозії металів, старіння полімерів, окиснення жирів. Частина з них є антиоксидантами – речовинами, які гальмують або запобігають окисненню органічних речовин і, перш за все, псуванню харчових продуктів. Так, для запобігання псуванню жировмісних і вітамінних продуктів додають вітамін Е, додециловий етер галлової кислоти, іонол та ін. У більшості випадків каталізатори застосовуються на носіях-підкладках, інертних в хімічних реакціях. Це селікагелі, азбест, вугілля, цеоліти та ін. Носій збільшує поверхню каталізатора, знижує чутливість каталізатора до каталітичних ядів (CS2, H2S, Cl2, Br2, P, As, Hg, Pb), а у випадку використання ферментних препаратів як каталізаторів введення носіїв (іммобілізація ферментів) дає змогу багаторазового застосування цих біологічних речовин у каталізі. Каталітичний процес відбувається у декілька стадій: наближення реагентів до каталізатора; адсорбція і орієнтація молекул реагентів на активних центрах поверхні каталізатора; деформація хімічних зв’язків у молекулах реагуючих речовин; хімічні перетворення активованих молекул; десорбція і видалення кінцевих продуктів реакції з поверхні каталізатора; регенерація каталізатора (у випадку отруєння). Прикладом гетерогенного каталізу є реакція дегідратації етанолу алюміній оксидом:
Мікрогетерогенний каталіз зв’язаний з ферментативними процесами. У рослинному, тваринному і людському організмі всі хімічні процеси каталізуються ферментами, які за своєю хімічною природою є білками з чітко спеціалізованими функціями. Білки утворюють псевдоколоїдні розчини, розмір частинок яких близький до колоїдних. За багатьма властивостями вони схожі з істинними розчинами: так, у білків відсутня межа поділу з розчинником, тому у розчинах, в яких відбуваються ферментаційні процеси, немає типової гетерогенності середовища. Такі процеси відносять до мікрогетерогенного каталізу. Ферменти входять до складу всіх клітин і тканин живих організмів. Вони обумовлюють здатність живих організмів здійснювати найрізноманітніші і в той же час цілком необхідні для життєдіяльності перетворення речовин. Сюди відносять процеси травлення білків, ліпідів, вуглеводів; використання речовин, які всмоктуються з кишкового тракту; вивільнення хімічної енергії, необхідної для всіх процесів життєдіяльності; поглинання кисню тканинами. Ферменти не тільки здійснюють розщеплення речовини, але й синтезують все те різноманіття органічних сполук, яке зустрічається у живих організмах. Таким чином, спектр дії ферментів йде від простих гідролітичних реакцій травного тракту аж до формування геному. Мікрогетерогенний або ферментативний каталіз відрізняється від гомогенного і гетерогенного каталізу принаймні за трьома параметрами: 1) висока специфічність ферментативного каталізу; 2) каталітична дія ферментів відбувається у порівнянно “м’яких” умовах (при температурі 37-40ºС, невисоких тисках (у межах 0,7 МПа) і рН); 3) висока каталітична активність ферментів. Наприклад, 1 моль заліза у складі каталази в 1 млрд. разів активніший, ніж 1 моль неорганічного заліза при розщепленні Н2О2 до Н2О і О2. §7. Основні властивості каталізаторів. Фактори, які впливають на каталіз. Важливою властивістю каталізаторів є відсутність їх впливу на величину константи рівноваги реакції. У зв’язку з цим каталізатор може тільки змінювати швидкість досягнення рівноваги, але не зміщувати її. Для каталізаторів характерною ознакою є специфічність. Каталізатори утворюють тимчасові проміжні сполуки з реагуючими речовинами. Для ефективної дії каталізатора необхідно, щоби він володів хімічною спорідненістю до реагенту. У цьому відношенні каталізатори володіють специфічною вибірковістю. Особливо яскраво вона проявляється у ферментів. Кожний фермент діє на відповідний субстрат чи на дуже обмежену їх кількість або на певний тип хімічного зв’язку у молекулі речовини. Так, наприклад, фермент цукраза гідролізує в цукрозі глюкозидний зв’язок між глюкозою і фруктозою і цей же зв’язок у молекулі трицукриду рафінози з утворенням дицукриду мелібіози і фруктози. Хоча деякі системи можуть реагувати і за декількома напрямками, каталізатори викликають прискорення процесу тільки у якому-небудь одному із можливих напрямків. Так, у реакції взаємодії карбон (ІІ) оксиду з воднем:
у присутності каталізатора міді при тиску 303,9-405,2 кПа утворюється метанол (а); якщо підвищити температуру до 550-600ºС, то метанол буде окиснюватися до метаналю (б); при цих же умовах введення лугу зумовлює утворення вищих спиртів (в); у присутності нікелю при 250ºС та нормальному тиску утворюється метан (г). Із метаналю при полімеризації одержують парафін (д). Тут проявляється вибірковість каталізатора – в реакційній суміші раніше буде відбуватися та реакція, яка вимагає меншої енергії активації. Зниження енергії активації. Швидкість хімічних реакцій залежить від числа зіткнень активних молекул. Частота таких зіткнень пов’язана з тривалістю існування активних молекул і їх концентрації. Каталітичні процеси відбуваються переважно через стадії утворення проміжних сполук реагенту з каталізатором, що супроводжується збільшенням швидкості хімічної реакції. Каталізатор у реакційному середовищі знижує енергію активації молекул реагуючих речовин, прискорюючи реакцію. Перебіг реакції А+В → АВ (а) вимагає затрати енергії активації Е1 у відсутності каталізатора. У присутності каталізатора К відбуваються дві наступні реакції: А+К → АК… (б) Енергія активації Е2 цієї реакції значно нижча від енергії активації реакції (а) (Е2 < Е1): АК+В → АВ+К… (в) Енергія активації Е3 у цій реакції нижча, ніж Е1 і Е2, тобто Е3 < Е2 (див. § 6). Таким чином, у присутності каталізатора процес відбувається з утворенням проміжних продуктів реакції АК. Найважливішою перевагою цього процесу є те, що сума енергії активації у реакціях (б) і (в) нижча, ніж в реакції (а), тобто Е2+Е3 < Е1. Зниження енергії активації не однакове при дії різних каталізаторів. Так, енергія активації розкладу пероксиду (Н2О2) без каталізатора 75000 кДж/моль (Е1), за участю колоїдної платини – 49140 кДж/моль (Е2), під впливом каталази печінки – 23100 кДж/моль (Е3) (рис. 5.2). Вплив дисперсності каталізатора. При гомогенному каталізі швидкість процесу пропорційна концентрації каталізатора. При гетерогенному ж каталізі важливе значення має величина питомої поверхні каталізатора. Оскільки величина питомої поверхні залежить від ступеня дисперсності, то його зміна різко відображається на активності каталізатора, а інколи – і на направленості реакції.
Рис. 5.2. Зниження енергії активації. Так, наприклад, ріст каталітичної активності платини залежить від збільшення поверхні каталізатора: платинова дротина < порошок < платинова чернь < колоїдна платина (золь). Фактори, які впливають на каталітичні процеси, по суті є тими ж, що і фактори, що впливають на швидкість хімічних реакцій. Вплив температури. Підвищення температури збільшує швидкість каталітичних процесів. Кожний каталізатор залежно від його властивостей проявляє максимальну активність при чітко визначеній температурі. Підвищення чи зниження температури негативно відображається на активності каталізаторів і це, перш за все, стосується екзотермічних процесів, при яких необхідне видалення надлишку тепла, та біологічних каталізаторів – ферментів, які в силу своєї білкової природи дуже чутливі до зміни температур. Вплив тиску. У деяких каталітичних реакціях швидкість значно змінюється при коливанні тиску, а окремі каталітичні реакції взагалі в умовах нормального тиску відбуватися не можуть (синтез вищих спиртів та ін.). Збільшення тиску прискорює хімічний процес внаслідок зростання числа ефективних зіткнень реагуючих частинок. Вплив розчинника на швидкість каталітичних реакцій особливо яскраво проявляється при гомогенному каталізі. Молекули полярних розчинників сприяють переходу молекул реагуючих речовин в активний стан. Під впливом молекул розчинника частинки реагуючих речовин стають більш реакційноздатними і переходять в активну йонізовану форму. Вплив активаторів та інгібіторів. Каталітична дія окремих каталізаторів підвищується або послаблюється присутністю у реакційному середовищі деяких речовин. Речовини, присутність яких сприяє швидкому перебігу каталітичної реакції, називаються активаторами. Ті речовини, які гальмують або припиняють дію каталізатора, називають інгібіторами. §8. Теорії каталізу. Для пояснення механізму каталізу було запропоновано велику кількість різноманітних теорій, окремі з яких доповнюють одна одну, а тому роглядати можна лише ті з них, які відображають основні напрямки у вченні про механізм каталізу: - теорія утворення проміжних сполук; - адсорбційна теорія; - електронна теорія; - мультиплетна теорія. Теорія проміжних сполук. Ця теорія базується на тому, що каталізатор у процесі реакції активно бере участь в утворенні нестійкої проміжної сполуки з реагентами. В результаті виникнення таких сполук знижується енергія активації хімічного процесу (див. §6). Проміжні сполуки при гетерогенному каталізі утворюються на поверхні каталізатора. В утворенні каталізатор-субстратного комплексу частинки субстрату зазнають деякої структурної деформації, яка супроводжується зміною міцності окремих хімічних зв’язків і в результаті цього зниженням енергетичного бар’єра, що обумовлює швидший перебіг хімічної реакції. Адсорбційна теорія каталізу. У цій теорії допускається, що швидкість хімічного процесу зростає при адсорбції молекул реагуючих речовин на частинах каталізатора. При цьому підвищується концентрація реагуючих молекул і збільшується можливість їх ефективних зіткнень, а також зростає їх реакційна здатність у результаті деформації адсорбованих молекул. Суть деформаційного процесу можна показати на прикладі дегідрування спирту на мідному каталізаторі:
При адсорбції на міді молекули спирту орієнтуються ОН-групами до поверхневих атомів металу. Відбувається деформація гідроксильної групи спирту, аж до розриву зв’язку між О-Н під дією активних центрів каталізатора і в результаті вивільнюється атом Гідрогену, а молекула спирту, втрачаючи ще атом Гідрогену, десорбується з поверхні каталізатора вже у вигляді альдегіду. Вважають, що при адсорбції молекули реагуючої речовини фіксуються на каталізаторі у крайньому випадку двома точками, в результаті чого між активними центрами відбувається деформація або розтягування зв’язків з наступним їх розривом або перерозподілом. Електронна теорія каталізу. В основі теорії лежить уявлення про те, що каталізатор має вільні або слабозв’язані електрони. Такими електронами забезпечуються вільні валентності на поверхні каталізатора, за рахунок яких адсорбуються молекули реагуючих речовин з утворенням вільних атомів і радикалів. При взаємодії вільних атомів і радикалів утворюються продукти реакції. Наприклад, безпосереднє здійснення реакції Н2+О2 → Н2О ускладнене через насиченість зв’язків реагуючих речовин. На платиновому каталізаторі вільні електрони переходять до молекули кисню, утворюючи йони кисню (2О–). На позитивно зарядженій платині адсорбуються молекули водню, віддаючи її електрони і перетворюючись у позитивно заряджені йони (2Н+). Йони Н+ і О–, взаємодіючи між собою, утворюють воду. Мультиплетна теорія каталізу. Згідно з нею на поверхні твердого каталізатора відбувається взаємодія реагуючих речовин з декількома атомами каталізатора (металу, його оксиду, сульфіду та ін.). Для каталізаторів характерна міцна кристалічна решітка, між атомами якої однакові відстані. Активний центр каталізатора найчастіше складається з двох, рідше – із декількох атомів (мультиплет). Відстань між атомами решітки – 2,48-0,277 нм. Каталіз відбувається після контакту (“накладки”) відповідних молекул реагуючої речовини з атомами каталізатора. Весь процес може бути представлений загальною схемою: вихідна речовина+каталізатор → мультиплетний комплекс → каталізатор+продукти реакції. Найяскравішим прикладом, що пояснює суть мультиплетної теорії, може бути реакція дегідрування циклогексану за участю, як каталізатора, платинового секстету:
С6H12 → C6H6+3H2 У цій реакції три атоми (1, 3, 5) каталізатора відтягують атоми гідрогену від усіх атомів карбону циклогексану з утворенням 3Н2, а три інших атоми платини (2, 4, 6) беруть участь в утворенні молекул бензену і трьох подвійних зв’язків між атомами карбону. Мультиферментна теорія має обмежене застосування, оскільки для вирішення практичних задач необхідно знати енергії зв’язків окремих атомів з каталізаторами, а вони невідомі.
§9. Ферментативний каталіз. Ферментами або ензимами називають біологічні каталізатори, які є продуктами життєдіяльності живих організмів. Так як і у технічних каталізаторів, у ферментів в гетерогенному каталізі бере участь не вся молекула, а лише окремі ділянки, які називаються активними центрами. Ферменти поділяються на дві групи: прості ферменти-білки і складні ферменти-білки. Перші є ферментами тільки білкової природи, а другі складаються з білка (апофермент) і небілкового компонента, яким можуть бути вітаміни, їх ефіри ортофосфатної кислоти, нуклеотиди, геми, йони металів. Небілкові компоненти складних ферментів називають простетичними групами, кофакторами, коферментами. Взаємодія між активними центрами ферментів і молекулами субстрату визначається силами хімічних, ковалентних, електростатичних зв’язків і до певної міри водневими зв’язками та вандерваальсовими силами. У складних ферментів функцію активних центрів виконують переважно простетичні групи із залученням білкових компонентів на окремих ділянках молекул білка. У простих ферментів активні центри утворюються за рахунок своєрідного розташування амінокислотних залишків у структурі білка. До таких амінокислотних залишків належать – SH-групи цистеїну; ОН-групи серину; NH-група імідазолу в гістидині, а також карбоксильні групи аспарагінової і глутамінової амінокислот, індольна група триптофану та ін. Суттєвим моментом, що визначає швидкість ферментативної реакції, є концентрація реагуючих речовин, враховуючи тим більше те, що фермент попередньо утворює проміжну сполуку з субстратом. При вивченні кінетики ферментативних процесів проводять вимірювання початкових швидкостей реакцій, що досягається шляхом зміни концентрації субстрату при незмінності всіх інших умов. Міхаеліс і Ментен розробили теорію, яка пояснює залежність початкової швидкості від концентрації субстрату. Вони виходили із такого рівняння: F+S де: F – фермент (ензим); S – субстрат; FS – фермент-субстратний комплекс; P – продукт реакції; k – константи реакцій. За умови, що [S]>>[F]o, i [S]o>>[FS] та [S]о ≈ [S] ([F]o і [S]o – початкові концентрації ферменту і субстрату, початкову швидкість νo утворення продукту можна описати рівнянням:
Концентрація ферменту і субстрату в момент часу τ від початку реакції: [F] = [F]0–[FS], (5.16) [S] = [S]0–[FS]. (5.17) Міхаеліс і Ментен припустили, що k–1>>k2. Тому першу стадію утворення комплексу [ES] можна розглядати як процес швидкого встановлення динамічної рівноваги. Константа рівноваги:
Якщо в рівняння (5.18) підставити значення [F] згідно з (5.16), то:
звідси:
На підставі (5.15) і (5.20) можна визначити υ0. Виходячи з умови, що [S]0>>[F]0, витратою субстрату при вивченні початкової швидкості можна знехтувати, тобто [S]0 ≈ [S], тому:
Часто величина k2 наближається до k–1 або навіть перевищує її. Тому був запропонований інший підхід, незалежний від відносних величин k–1 і k2, за умови перебігу реакції у стаціонарному режимі, тобто якщо
Це дає можливість записати кінетичне рівняння реакції:
k1([F]o–[FS])[S]–(k–1+k2)[FS] = 0. (5.22) Тоді відносно [FS], одержимо:
Відповідно рівнянь (5.15) і (5.23) вираз для початкової стаціонарної швидкості буде таким:
де Km – константа Міхаеліса-Ментена яка дорівнює:
Рівняння (5.24) є рівнянням Міхаеліса-Ментена. Рівняння (5.21) є окремим випадком рівняння Міхаеліса-Ментена, коли k–1>>k2, тому:
Добуток k2·[F]o має розмірність швидкості реакції, що звичайно називають швидкістю ферментативної реакції і позначають υm (υm – швидкість, з якою відбувається реакція, якщо фермент перебуває у складі комплексу FS). Тобто, при [F]o = [FS], або υm = k2[F]0. (5.26) Тоді рівняння (5.24) можна записати так:
Отже, стаціонарна швидкість ферментативної реакції (5.14) при [S]0>>[F]0 має гіперболічну залежність від концентрації субстрату і лінійну – від початкової концентрації ферменту (рис. 5.3 а, б).
а) б) Рис. 5.3. Залежність початкової швид-кості υ0 ферментативної реакції від концентрацій субстрату (а) і ферменту (б). Ці залежності дійсно характеризують кінетику більшості ферментативних реакцій, а рівняння Міхаеліса-Ментена формально описує початкову стаціонарну швидкість. Рівняння (5.27) має два граничних випадки. При низькій концентрації субстрату Кm>>[S] швидкість описується рівнянням першого порядку відносно [S]: Обчислення Кm спрощується, якщо використати замість рівняння (5.27) його зворотні показники, згідно з Лайнуівером-Берком і за допомогою графіка (рис. 5.4) можна визначити Кm, оскільки при
Рис. 5.4. Графічне визначення υm і Кm заЛайнуівером-Берком. Таким чином, константа Міхаеліса Кm чисельно дорівнює концентрації субстрату, при якій швидкість ферментативної реакції досягає половини максимальної υm. Визначення ферментативної активності використовується з діагностичною метою. Наприклад, інфаркт міокарда супроводжується різким підвищенням активності ферменту аспартатамінотрансферази (АсАТ) ще до появи електрокардіографічних змін у міокарді. Тому метод діагностики за активністю АсАТ точніший і дає відомості раніше.
КОНТРОЛЬНІ ЗАВДАННЯ 1. Що розуміють під швидкістю реакції? 2. Чим визначається молекулярність реакції? 3. Сформулювати закон діючих мас. 4. Що таке константа швидкостей реакції? 5. Написати вирази швидкості реакції першого, другого, третього і нульового порядків. 6. Як залежить швидкість реакції від температури? Що таке температурний коефіцієнт? 7. Які основні положення теорії активних зіткнень? 8. Паралельні і послідовні реакції. 9. Оборотні і спряжені реакції. Навести приклади. 10. Дати визначення енергії активації. 11. Гомогенний каталіз. Привести приклад. 12. Гетерогенний каталіз. Поняття про інгібітори. 13. Як залежить швидкість реакції від концентрації реагуючих речовин? 14. Поняття про мікрогетерогенний каталіз. 15. Що лежить в основі дії каталізаторів? 16. Вплив температури, тиску, активаторів та інгібіторів на каталітичну дію. 17. Назвати основні теорії каталізу. 18. Теорія проміжних сполук. 19. Адсорбційна теорія каталізу. 20. Електронна і мультиплетна теорія каталізу. 21. Що таке ферментативний каталіз? 22. До якого класу речовин відносять ферменти? 23. Особливості дії ферментів як каталізаторів. 24. Які фактори впливають на активність ферментів?
ЧАСТИНА ДРУГА КОЛОЇДНА ХІМІЯ
Розділ VІ АДСОРБЦІЯ Вчення про адсорбцію почало інтенсивно розвиватися у кінці XIX і на початку XX століття, хоча вперше адсорбцію газів вугіллям виявив італієць Ф.Фонтана ще у 1777 році, а через 7 років акад. Г.Ловіц використав явище адсорбції вугіллям для очищення розчинів. Адсорбція (від лат. ad – на, при і sorbeo – поглинаю) – явище накопичення однієї речовини на поверхні іншої. Накопичення ж її всередині іншої речовини називають абсорбцією. Речовина, яка адсорбується, називається адсорбтивом; речовина, на поверхні якої відбувається адсорбція – адсорбентом. Адсорбція є зворотним процесом. Процес зворотний до адсорбції, називається десорбцією. Видалення адсорбованих речовин з адсорбентів за допомогою розчинників називають елюцією. Розрізняють молекулярну і йонну адсорбцію, залежно від того, що адсорбується – молекули чи йони речовини. При адсорбції може відбуватися хімічна взаємодія адсорбента з адсорбтивом, наприклад: СаО+СО2 → СаСО3. адсорбент адсорбтив Така адсорбція називається хемосорбцією. Процес адсорбції залежить від фізичної і хімічної природи адсорбента й адсорбтива. Так, наприклад, на активованому вугіллі краще адсорбуються ароматичні сполуки, ніж аліфатичні. Часто адсорбція підвищується зі збільшенням числа подвійних зв’язків в адсорбтиві.
§1. Адсорбція на поверхні рідин. На поверхні рідин можуть адсорбуватися частинки речовин, що розчинені у рідинах. Адсорбція супроводжує процес розчинення, впливаючи на розподіл частинок розчиненої речовини між поверхневим шаром розчинника і внутрішнім його об’ємом. На основі другого закону термодинаміки величина поверхневої енергії рідин зменшується до мінімуму. У чистих розчинниках зменшення цієї енергії відбувається шляхом зменшення поверхні. У розчинах поверхнева енергія може знижуватися або зростати за рахунок зміни концентрації частинок у поверхневому шарі рідини. Гіббс встановив, що розподіл розчиненої речовни відбувається таким чином, щоб досягалося максимальне зниження поверхневого натягу. Ним же запропоноване рівняння залежності величини адсорбції Г від концентрації розчиненої речовини на певному об’ємі поверхневого шару порівняно з таким же об’ємом всередині рідини:
де: Г – адсорбція, молярний надлишок (недостача) розчиненої речовини на 1 м2 поверхні, моль/м2; c – загальна концентрація розчину, моль/м2; R – газова стала 8,314 Дж/моль·K; Т – абсолютна температура, K (273+t°C); Рівняння Гіббса є математичним обгрунтуванням загального правила: речовина, яка зменшує поверхневий натяг, концентрується у поверхневому шарі, і навпаки. Якщо поверхневий натяг зменшується при збільшенні концентрації розчиненої речовини, то Метою негативної адсорбції є повне витіснення адсорбтиву із поверхневого шару всередину об’єму розчинника. В результаті різниці концентрацій виникає дифузія, яка буде направлена у поверхневий шар. Тому у поверхневому шарі буде деяка кількість адсорбтиву. Речовини, які різко підвищують поверхневий натяг, майже повністю відсутні у поверхневому шарі розведених розчинів. Лише значне збільшення концентрації таких розчинів призводить до переміщення у поверхневий шар помітних кількостей розчиненої речовини, що супроводжується збільшенням поверхневого натягу. Речовини, що негативно адсорбуються, називаються поверхнево-інактивними. До них належать мінеральні солі, вуглеводи та інші сполуки. Позитивну і негативну адсорбцію можна пояснити на прикладі адсорбції масляної кислоти у воді. Масляна кислота володіє меншим поверхневим натягом, ніж вода (σмасл.кислоти = 27 ерг/см2 а σводи = 72,75 ерг/см2 при 20ºС). Перші порції масляної кислоти, доданої до води, розподіляються майже виключно у поверхневому шарі, різко знижуючи поверхневий натяг. Наступні порції кислоти вже викликають дифузію у нижчі шари води. Зміна поверхневого натягу води продовжується з поступовим уповільненням аж до величини 27 ерг/см2, тобто до значення поверхневого натягу масляної кислоти. Це буде означати, що поверхневий шар розчину складається тільки з молекул масляної кислоти. Якщо приливати воду до кислоти, то процес адсорбції буде змінюватися у зворотному порядку. Перші порції води адсорбуються негативно, практичного не впливаючи на величину поверхневого натягу кислоти. Подальше додавання води призводить до збільшення числа її молекул у поверхневому шарі і до прогресуючого збільшення поверхневого натягу (рис. 6.1.).
Рис. 6.1. Зміна по-верхневого натягу водного розчину масляної кислоти. Позитивна і негативна адсорбції мають велике значення для обміну речовин у живих організмах. Поверхневий натяг біологічних рідин (табл. 6.1) значно менший, ніж води завдяки присутності в них поверхнево-активних речовин. Тому гідрофобні речовини, наприклад кислоти жирного ряду, стероїди, будуть накопичуватися біля стінок судин, клітинних мембран, що полегшує їх проникність крізь ці мембрани та обмінні процеси. Для адсорбції із водних розчинів велике значення має наявність у молекул речовини полярних (гідрофільних) і неполярних (гідрофобних) груп. Так, молекула масляної кислоти містить групу СООН і гідрофобний вуглеводневий ланцюг: СН3–СН2–СН2–. Таблиця 6.1. Поверхневий натяг деяких рідин на межі поділу з повітрям. Рідини Поверхневий натяг, ерг/см2 Ртуть Вода Гліцерол Сироватка крові (при 38ºС) Етанол Етер 430,0 72,7 65,0 46-47 22,0 16,0
Молекули, які володіють одночасно обома видами груп, називаються дифільними. У дифільних молекул з коротким гідрофобним ланцюгом переважають гідрофільні властивості, тому такі молекули добре розчиняються у воді і для них є характерна негативна адсорбція. З видовженням вуглеводнего ланцюга посилюються гідрофобні властивості молекул й понижується їх розчинність у воді. Отже, до поверхнево-активних належать речовини дифільної структури, які мають менший, ніж розчинник, поверхневий натяг і розчинення яких призводить до позитивної адсорбції, викликаючи зниження поверхневого натягу. Поверхнево-інактивні речовини володіють протилежними властивостями. Одночасно зі збільшенням гідрофобних властивостей молекул зростає їх поверхнева активність. Так, видовження ланцюга в гомологічному ряді жирних кислот, спиртів, амінів та ін. на радикал –СН2– підвищує їх здатність до позитивної адсорбції у розведених розчинах у 3-3,5 раза (правило Траубе-Дюкло). Молекули речовини з перевагою гідрофобних властивостей (жирні кислоти з великою молекулярною масою та ін.) розташовуються на поверхні води так, що утворюють поверхневі плівки. При незначних кількостях таких молекул поверхнева плівка не утворюється. Якщо ж молекул багато, то вони розташовуються упорядковано, одна біля одної, причому їх гідрофобні частини виступають над водною поверхнею, утворюючи так званий частокол Ленгмюра (рис. 6.2).
Рис. 6.2. Поверхневі плів-ки: 1 – хаотичне розташу-вання дифільних молекул; 2 – частокол Ленгмюра; 3 – надлишок молекул; 4 – гідрофільна частина моле-кул; 5 – гідрофобна части-на молекул. Поверхнева пліва утворюється мономолекулярним шаром молекул, кожна з яких займає на поверхні води відповідну площу, яку можна розрахувати, виходячи з того, що у випадку насиченої адсорбції і утворення мономолекулярної плівки на 1 см2 поверхневого шару адсорбується Г∞ моль речовини, тобто Г∞Na молекул (Na – стала Авогадро). Отже, площа S0, яку займає одна молекула, рівна:
Товщину шару (довжину молекули) можна знайти, розрахувавши масу речовини на 1 см2, яка дорівнює: m = υd = 1ld, (6.3) де: l – довжина молекули; d – густина речовини. Але з іншого боку маса речовини рівна: m = Г∞M, (6.4) тобто, добутку величини максимальної адсорбції на молярну масу. Порівнявши вирази (6.3) і (6.4), можна вирахувати довжину молекули l:
Наприклад, молекули жирних кислот з однією полярною групою (масляна, валеріанова, капронова і т. п.) займають на поверхні води площу 21·10–16 см2 незалежно від довжини вуглеводневого ланцюга, тоді як кислоти з двома полярними групами (олеїнова) займають площу удвічі більшу, а молекули з трьома полярними групами (наприклад, тристеарин) – у три рази більшу площу (табл. 6.2).
Таблиця 6.2. Залежність площі, яку займає на поверхні води, від числа полярних груп у молекулі. Речовина Формула Полярна група Число Площа одної молекули, см2 Площа одної полярної групи, см2 Пальмітинова к-та Стеаринова к-та Олеїнова к-та
Тристеарин
Триолеїн С15Н31СООН С17Н35СООН С17Н33СООН
(С18Н35О2)3С3Н5
(С18Н33О2)3С3Н5 Карбоксил
Карбоксил
Карбоксил і подвійний зв’язок
Складноетерні зв’язки
Складноетерні і подвійні зв’язки
21·10–16 22·10–16 44·10–16
66·10–16
126·10–16 21·10–16 22·10–16 22·10–16
22·10–16
21·10–16
Утворення поверхневих плівок ускладнює процес фільтрації. На межі повітря-вода бульбашками повітря, що є в розчині, може адсорбуватися поверхнево-активна речовина. Плівка цієї речовини утворює оболонку навколо бульбашки (рис. 6.3). Така бульбашка при проштовхуванні через пори у фільтрі не здатна до різкої деформації і тому може закупорювати більші отвори у фільтрі, ніж бульбашки без плівки. У скафандри водолазів повітря подається під тиском і, відповідно, у крові водолаза розчиняється підвищена кількість газів. Надто швидке підняття на поверхню призводить до різкого зниження тиску у скафандрах і значна частина газів крові видаляється у вигляді бульбашок, на яких утворюється поверхнева плівка поверхнево-активних речовин крові. Бульбашки газів закупорюють дрібні судини у різних тканинах і органах, що призводить до кесонної хвороби з важкими наслідками аж до смерті.
Рис. 6.3. Поверхнева плів-ка навколо повіт-ряної бульбашки. Така ж хвороба виникає в результаті різкого зниження атмосферного тиску при розгерметизуванні скафандрів льотчиків і кабін літаків при висотних польотах. Лікування кесонної хвороби здійснюється поміщенням хворого в барокамеру, де задається підвищений тиск. Бульбашки газів знову розчиняються у крові і при наступному уповільненому (протягом декількох діб) зниженні тиску у барокамері надлишок газів видаляється із крові через легені. §2. Адсорбція твердими тілами. Твердими тілами можуть адсорбуватися гази (в тому числі і пара) та молекули і йони розчинених речовин. Така адсорбція пояснюється наявністю силових полів притягання, що виникають за рахунок незрівноважених зв’язків у кристалічній решітці (рис. 6.4). На виступаючих ділянках твердого адсорбенту адсорбція відбувається особливо інтенсивно.
Рис. 6.4. Виступи або піки поверхні (за Тейлором). Так, виступи (активні центри) на частинці вугілля у 4,5 раза інтенсивніше адсорбують кисень, ніж заглиблення на його поверхні. Адсорбційні сили складаються із валентних сил взаємодії (хімічних) і слабших вандерваальсових (фізичних). Їх роль у кожному випадку адсорбції різна. На самому початку адсорбції газів, коли тиск низький, спостерігається хімічна адсорбція; зі зростанням тиску хімічна адсорбція поступається місцем фізичній, яка в основному визначає адсорбцію газів. Процес адсорбції є оборотним. Адсорбовані частинки не залишаються нерухомими: вони утримуються на адсорбенті лише соті і тисячні частки секунди і десорбуючись, заміщуються новими частинками, до того ж вони не є чітко зафіксованими на адсорбенті і можуть мігрувати по його поверхні. Накінець встановлюється динамічна адсорбційна рівновага між вільними і адсорбованими частинками. Швидкість встановлення адсорбційної рівноваги різних речовин неоднакова; наприклад, при адсорбції СО2 на вугіллі рівновага настає через 20 сек, при адсорбції О2 – через 2,5 год, при адсорбції N2 – через 20 год. Швидкість адсорбції відіграє важливу роль. Так, у протигазі повітря дуже швидко, проходячи через адсорбційну обробку, очищується від домішок, а це можливо лише при високій швидкості адсорбційних процесів. При підвищенні температури знижується фізична адсорбція, тому що при посилюється рух молекул у адсорбційному шарі, порушується орієнтація адсорбованих молекул, тобто зростає десорбція. Але підвищення температури збільшує енергію адсорбованих частинок, що, згідно з теорією активації, посилює хімічну адсорбцію. Отже, в одних випадках підвищення температури посилює десорбцію, в інших – посилює адсорбцію. Для більшості газів підвищення температури знижує адсорбцію, у той же час підвищення температури від –185 до +20ºС у 10 разів збільшує адсорбцію кисню платиною, оскільки при цьому зростає хімічна адсорбція. Хімічна адсорбція (хемосорбція) зумовлена хімічними реакціями між поверхневими молекулами адсорбенту і адсорбтиву. В результаті хемосорбції на поверхні утворються хімічні сполуки. Тепловий ефект близький до енергії утворення хімічних сполук – 40-120 кДж/моль, підвищення температури сприяє хемосорбції. Підвищення тиску газів і пари посилює адсорбцію. При адсорбції пари спостерігається так звана капілярна конденсація. Сконцентрована в капілярах рідина утворює ввігнутий меніск, над яким пара є насиченою при більш низькому тиску, ніж над плоскою поверхнею. Це підвищує конденсацію пари у капілярах адсорбента.
§3. Адсорбція із розчинів. При адсорбції із розчинів разом з розчиненою речовиною адсорбуються молекули розчинника. Кількість адсорбованих частинок розчиненої речовини і розчинника залежить від їх власної адсорбційної здатності та концентрації розчину. При малих концентраціях переважає адсорбція розчиненої речовини, а при високих – адсорбція розчинника. Залежність адсорбції
(де С0 – концентрація розчиненої речовини до адсорбції; С – концентрація речовини після адсорбції; m – маса адсорбенту) від концентрації адсорбтиву у розчині можна зобразити графічно (рис. 6.5).
Рис. 6.5. Ізотерма адсорб-ції із розчину. Спочатку кількість адсорбованої речовини зростає зі збільшенням її концентрації у розчині, але потім починає переважати адсорбція розчинника і концентрація речовини у розчині підвищується, тому С0<C<0, і дріб
стає від’ємним (крива опускається нижче осі абсцис). Отже, молекули розчинника, адсорбуючись на поверхні твердого адсорбенту, зменшують адсорбційність адсорбтиву. Наприклад, розчин ацетатної кислоти в толені. Толуен краще, ніж ацетатна кислота, адсорбується активованим вугіллям. Концентрація кислоти у процесі адсорбції зростає. При адсорбції рідкої речовини твердим адсорбентом виділяється теплота, яка називається теплотою змочування і визначається кількістю джоулів, що виділяється при адсорбції 1 г твердого адсорбенту рідини. Різні адсорбенти володіють неоднаковою теплотою змочування (табл. 6.3). Таблиця 6.3. Теплота змочування різних адсорбентів (за А.Думанським). Адсорбент Теплота змочування, Дж/г води бензену Силікагель Вугілля Ґрунт Торф 81,22 48,56 39,77 61,96 - 118,48 12,14 10,88
Ці дані вказують, що найбільшою адсорбційною здатністю до води володіє силікагель, а до бензолену – активоване вугілля. П.Ребіндер назвав адсорбенти, які інтенсивно взаємодіють з водою, гідрофільними, а ті, що погано – гідрофобними. Отже, гідрофільним адсорбентом є силікагель, гідрофобним – вугілля. Це явище відіграє суттєву роль для плодючості ґрунтів. Залежно від змочуваності водою ґрунти поділяють на гідрофільні і гідрофобні. Теплота змочування впливає на температурний режим ґрунтів. У практиці виникає потреба у підвищенні або пониженні гідрофільності чи гідрофобності поверхні твердого тіла. Для гідрофобізації поверхні як змочувальні реагенти використовують розчини будь-якої жирної кислоти. При цьому молекули кислоти, адсорбуючись на твердій поверхні, утворюють орієнтований шар, в якому полярні групи молекул повернуті до поверхні матеріалу, вуглеводневі ланцюги – у розчин. Поверхня речовини набуває гідрофобних властивостей. Краплі води на такій поверхні утворюють тупі крайові кути (рис. 6.6). Жировими речовинами обробляють поверхню різноманітного устаткування й інвентаря, щоб запобігти їх замочування водою.
Рис. 6.6 Гідрофобізація поверхні: а – гідрофільна поверхня; б – поверхня, гідрофобізована поверхнево-актив-ною речовиною. Іноді, навпаки, гідрофобним поверхням потрібно надати властивості гідрофільності. Наприклад, шкіра рук, білизна, посуд внаслідок забруднення жировими речовинами втрачають властивість змочуватися водою, стають гідрофобними. Якщо обробити їх милами, вони знову стають гідрофільними. При адсорбції із розчинів, що містять суміш багатьох речовин, в одних випадках речовини адсорбуються у пропорційних кількостях до адсорбційної здатності кожної з них, в інших випадках одні речовини запобігають адсорбції інших (антагонізм); нарешті, можливі випадки, коли речовини взаємно посилюють адсорбцію (синергізм).
§4. Фізико-математичне обгрунтування адсорбції. На межі поділу газ-рідина величини адсорбції можна визначити за зміною поверхневого натягу. Для цього визначають поверхневий натяг при постійній температурі для розчинів різних концентрацій і будують ізотерму поверхневого натягу. Наприклад, при адсорбції масляної кислоти на поверхні води, її поверхневий натяг зменшується залежно від кількості адсорбованої кислоти (рис. 6.7), у той же час ізотерма адсорбції прямує до межі, якою є повне насичення поверхневого шару частинками адсорбованої речовини. Користуючись ізотермою поверхневого натягу, за допомогою рівняння Гіббса (6.1) можна розрахувати кількість адсорбованої речовини (рис. 6.8).
Рис. 6.7. Ізотерми поверх-невого натягу і адсорбції масляної кислоти. Для концентрації СА величину поверхневої активності Вираховувати величину адсорбції за зміною поверхневого натягу в рідинах зручно, але тільки для поверхнево-активних речовин, бо вони можуть різко змінювати поверхневий натяг. Ленгмюр на основі передбачення про мономолекуляний адсорбційний шар ввів універсальніше рівняння ізотерми адсорбції.
Рис. 6.8. Графічне визначення по-верхневої активності. Наприклад, активні центри А на поверхні адсорбенту взаємодіють з молекулами газу М, причому кожний центр зв’язує лише одну молекулу газу. Взаємодію між ними можна записати так: A+M Тоді: ν1 = K1[A][M]; ν2 = K2[AM]. При адсорбційній рівновазі ν1 = ν2, або K1[A][M] = K2[AM]. Розділивши обидві частини рівняння на К1[AM], одержимо:
де Допустимо, що можлива концентрація молекул газу 1 см2 поверхні адсорбенту за умови її повного насичення виражається через Г∞, через С – рівноважна концентрація газу, тобто концентрація газу, який залишився вільним після настання адсорбційної рівноваги, через ГАМ – поверхнева концентрація адсорбованого газу (при адсорбційній рівновазі); ГА – концентрація молекул газу, яка додатково може приєднатися до адсорбенту (після настання адорбційної рівноваги) так, щоб була досягнута допустимо можлива концентрація молекул Г∞, тоді: ГА = Г∞–ГАМ. Позначення А, М, АМ у рівнянні (6.6) відповідають значенням концентрації газу, тобто: [M] = C; [A] = ГА; [AM] = ГАМ. Підставивши ці значення у рівняння (6.6), одержимо: КГ = (Г∞–Г)С; КГ = СГ∞–СГ; КГ+СГ = СГ∞; Г(К+С) = СГ∞;
Рівняння адсорбції Ленгмюра дозволяє проводити розрахунок кількості речовини, адсорбованої на одиницю поверхні адсорбенту. При низьких концентраціях речовини із знаменника рівняння (6.7) можна вилучити величину С, тоді:
тобто кількість адсорбованої речовини прямопропорційна її концентрації. При великих концентраціях речовини можна, навпаки, знехтувати відносно малим значенням К , тоді:
і після скорочення Г = Г∞, тобто, при великих концентраціях, адсорбується максимально допустима кількість речовини. Рівняння Ленгмюра відповідає ізотермі адсорбції (рис. 6.9). Г характеризує поверхневий надлишок, тобто різницю концентрацій адсорбованої речовини у поверхневому шарі і такому ж за товщиною шарі всередині рідини після настання адсорбційної рівноваги. Г∞ – допустимо можливе значення поверхневого надлишку; С – концентрація адсорбтиву після настання адсорбційної рівноваги. Зі збільшенням концентрації адсорбтиву зростає і кількість адсорбованої речовини, прямуючи до Г∞.
Рис. 6.9. Ізотерма адсорб-ції Ленгмюра. Висновок рівняння Ленгмюра базується тільки на існуванні мономолекулярного шару адсорбованої речовини. Однак адсорбція може бути і багатошаровою, тоді рівнянням ізотерми адсорбції Ленгмюра користуватися не можна. Виявилося, що більш точними є так звані S-подібні ізотерми Брунауера, Еммета і Теллера (скорочено БЕТ) (рис. 6.10). Ізотермі адсорбції Ленгмюра відповідає лише нижня частина (ОА) кривої. S-подібні ізотерми (і відповідні їм рівняння) особливо часто застосовуються для випадків адсорбції пари на твердих адсорбентах.
Рис. 6.10. S- подібна ізо-терма адсорбції БЕТ. Вивчаючи адсорбцію із розчинів, Фрейндліх запропонував емпірично виведене рівняння:
де: х – кількість адсорбованої речовини (адсорбату); m – маса адсорбенту; р – рівноважний тиск; с – рівноважна концентрація; Рівняння (6.8) відповідає ізотермі адсорбції Фрейндліха (рис. 6.11). Для визначення констант рівняння Фрейндліха логарифмують і одержують рівняння прямої:
Рис. 6.11. Графічне визначен-ня констант у рівнянні Фрейндліха Графічна залежність lgГ від lgс виражається прямою (рис. 6.11), що відсікає по осі ординат відрізок ОА, який рівний lgK (при с = 1, Г = K, lgс = 0 і lgГ = lgK). Тангенс кута φ – нахилу прямої до осі абсцис дає значення другої константи
§5. Вибіркова адсорбція і її біологічне значення. Якщо адсорбція будь-якої речовини значно переважає адсорбцію інших, то можна говорити про її вибіркову адсорбцію цим адсорбентом. Ця обставина має велике практичне значення. Підбираючи потрібні адсорбенти, можна відділити із складних сумішей чітко визначену речовину. Прикладом вибіркової адсорбції є йонна. Згідно з правилом Панета-Фаянса, на твердому адсорбенті переважно адсорбуються йони, які входять до складу адсорбенту, або містять спільну з адсорбентом групу. Так, на частках AgCl, що утворюються у реакції AgNO3+KCl, адсорбуються або йони Cl–, або Ag+, але не K+ чи NO–3. Багатовалентні йони адсорбуються сильніше, ніж одновалентні. Йони однакової валентності також адсорбуються неоднаково у зв’язку з відмінностями їх ступеня гідратації. За їх здатністю до адсорбції вони розташовуються у так звані ліотропні ряди: Cs+>Rb+>K+>Na+>Li+; CNS–>J–>NO–3>Br–>Cl–. Ці особливості йонної адсорбції мають велике значення для процесів стабілізації і коагуляції колоїдів. В організмі людей і тварин часто спостерігається явище вибіркової адсорбції токсинів та інших речовин різними тканинами і клітинами. Так, наприклад, токсини збудників дизентерії вражають перш за все вегетативну нервову систему, а токсини збудників ботулізму – центральну нервову систему; сипний тиф вражає стінки судин шкіри, мозку, серця. Імунні білки (антитіла) володіють високою вибірковістю у сполученні тільки з чітко визначеним для кожного антитіла чужорідним білком (антигеном). За допомогою електронної мікроскопії було встановлено, що антитіла адсорбуються на поверхні бактерій черевного тифу не рівномірно, а ділянками, ніби “активними центрами”. Навіть невеликі кількості введених в організм токсинів, що володіють високою адсорбційною здатністю на активних центрах окремих ферментів і інших біологічно активних сполук, призводять до їх блокади. Зокрема, введення в організм ціанистих сполук викликає миттєву смерть через блокаду ферментів цитохромної системи дихального ланцюга. Явище адсорбції використовується у так званій адсорбційній терапії, яка полягає у введенні хворому адсорбентів для поглинання шкідливих речовин. Активоване вугілля (карболен), аеросил (аморфний SiO2) використовують для зв’язування ядів, токсинів, які потрапили у шлунково-кишковий тракт, та для адсорбції кишкових газів.
§6. Обмінна адсорбція. Під поняттям обмінної адсорбції слід розуміти явище заміщення на адсорбенті одної речовини на іншу. Найпростіший випадок – витіснення слабкого адсорбтиву сильнішим; наслідком “конкуренції” за активні центри адсорбенту на ньому будуть обидва адсорбтиви у кількостях, пропорційних їх здатності до адсорбції. Отож, якщо адсорбція одного із адсорбтивів на даному адсорбенті у 10 разів вища, ніж у другого, то його й адсорбується у 10 разів більше. Однак, конкуруючі речовини впливають на зміну пропорційності їх адсорбції, тобто спостерігаються явища синергізму і антагонізму. Поширеним різновидом обмінної адсорбції є йонообмінна адсорбція. Її суть полягає у тому, що деякі адсорбенти містять хімічні групи, які дисоціюють і заміщують свої йони на однойменно заряджені йони, присутні у розчині (рис. 6.12). Якщо із розчину обмінюються йони Н+ чи ОН– (рис. 6.13), то в результаті обміну в розчині змінюється співвідношення цих йонів, а це призводить до зміни рН. Так, колоїдні частки Fe(OH)3 можуть обмінювати адсорбовані йони Cl– на ОН–, що веде до переваги в розчині йонів Н+ і, як наслідок, рН розчину змінюється у кислу зону. Такий випадок йонообмінної адсорбції називається гідролітичною.
Рис. 6.12. Схема йонообмінної адсорбції. Для науково-дослідної роботи і у промисловості використовуються йонообмінні смоли. Це зшиті нерозчинні полімери, які володіють властивостями кислот, основ і солей. До їх структури введені кислотні або основні угрупування, з якими зв’язані рухливі протийони, що здатні до обміну. Матрицю йонообмінних смол одержують полімеризацією або поліконденсацією. При цьому зв’язуючими лінійні полімерні молекули містокоутворюючими агентами найчастіше виступають при полімеризації дивінілбензен або дивініл, а при поліконденсації – формальдегід. Структурні схеми найрозповсюдженіших йонообмінних смол можна представити у вигляді:
де радикали R1 і R2 є сульфатні, карбоксильні, фосфатні або основні (амінні) групи. Всі йонообмінні смоли поділяються на аніоніти і катіоніти, залежно від виду обмінних йонів: аніонів чи катіонів. До катіонітів належать амберліт IR-100, Дауекс-50 та ін. Прикладом є синтетичні смоли фенолформальдегідного й інших типів з амінними групами. Це Дауекс-2 IRA-400, Вофатит-М та ін.
Рис. 6.13. Схема гідролітичної адсорбції. Функціональними групами катіонітів є карбоксильні, гідроксильні і сульфогрупи, у яких може обмінюватися катіон (гідроген йони заміщується на йон металу). Функціональні групи аніонітів (аміногрупи) обмінюють аніони, наприклад ОН– заміщується на Cl–, SO42– і т.п. §7. Хроматографія. Хроматогафічний (від грецьк. chromatos – фарба і chrapho – пишу) аналіз – це фізико-хімічний метод розділення речовин за допомогою сорбційних процесів при напрямленому русі однієї з фаз. В його основу покладені різні за механізмом та неодноразові повторення явищ сорбції та десорбції. Метод розробив М.Цвіт (1903). Ним був сконструйований прилад, що складався з лійки і колби (рис. 6.14). Скляна лійка заповнювалася безбарвним адсорбентом і через неї пропускалася водяна суміш пігментів, одержаних механічним подрібненням зелених листків. Проходячи крізь шар адсорбенту, різні речовини розміщувалися у вигляді окремих забарвлених зон. Найбільші частки адсорбтиву затримувалися верхніми шарами адсорбенту, а менші – середніми, найдрібніші виявлялися в нижніх шарах. Адсорбент і адсорбтиви висушувалися і розділялися на окремі зони. Для одержання в чистому вигляді окремих речовин вони вимивалися (елюювалися) відповідними розчинниками. Так були одержані хлорофіли а, в і с та ізомери ксантофілу. Забарвлений стовпчик адсорбенту і адсорбтиву було названо хроматограмою, а метод – хроматографією. У 1941 році А.Мартін і Р.Стінг розробили метод розподільної хроматографії на папері і використали його для одержання у чистому вигляді білків, амінокислот, вуглеводів та інших речовин.
Рис.6.14.Адсорбційна хроматографія (за Цвітом): 1 – розчин-ник, 2 – адсорбент з шарами адсорбованих речовин, 3 – вата, 4 – відведення до вакуумної установки. За механізмом процесу розділення хроматографія поділяється на такі види: адсорбційна, розподільна, екстракційна, йонообмінна, ексклюзивна (гельпроникна). Адсорбційна хроматографія грунтується на різній адсорбції тієї чи іншої речовини адсорбентом. Прикладом розподільної хроматографії є хроматографічний аналіз амінокислотного складу на папері (рис. 6.15). Волокна целюлози паперу адсорбують воду, і розділення речовин на ньому проходить між незмішуваним з водою розчинником і водним шаром волокон паперу. Цей вид хроматографії застосовується для розділення білків, вуглеводів, стероїдів, пуринів, фенолів, вітамінів, антибіотиків та ін. В основі йонообмінної хроматографії лежать різниці констант йонообмінної рівноваги між нерухомою фазою (йонітом) і компонентами суміші, що розділяється. Ексклюзивна хроматографія базується на різній проникності молекул компонентів суміші у нерухому фазу – високопористий гель. Прикладом може бути гель-фільтрація, коли суміш білка та інших речовин пропускають через гель сефадексу. Білок має високу молекулярну масу і тому швидше проходить через колонку, а молекули інших речовин розподіляються в порах сорбента заповнених розчинником. Екстракційна хроматографія здійснюється на основі неоднакової здатності компонентів рухомої фази (суміш, що розділяється) випадати в осад на твердій нерухомій фазі.
Рис. 6.15. Розділювальна хроматографія амінокислот на папері. Залежно від середовища, де відбувається розділення суміші, розрізняють газову і рідинну хроматографію. Газова хроматографія проводиться шляхом перенесення газом-носієм, під відповідним тиском, через колонку суміші компонентів. Відповідні фізичні прилади (рис. 6.16) фіксують величину сигналу, яка пропорційна концентрації речовини і відображається у вигляді відповідних хроматограм. За агрегатним станом нерухомої фази газова хроматографія буває газоадсорбційною (нерухома фаза – твердий адсорбент) і газорідинна (нерухома фаза – рідина). Рідинна хроматографія поділяється на рідинно-адсорбційну (твердорідинну) і рідинно-рідинну. За методом проведення розрізняють колонкову, тонкошарову, паперову і капілярну. У першому випадку використовуються спеціальні хроматографічні колонки, які заповнюються адсорбентом і крізь них пропускається рухома фаза, яка рухається внаслідок перепаду тиску. Різновидом колонкової хроматографії є капілярна, при якій твердий адсорбент наноситься твердим шаром на стінку капілярної трубочки. Тонкошарова хроматографія проводиться на площині відповідного сорбенту, що нанесений на металеву або скляну пластинку. У паперовій хроматографії як сорбент використовується спеціальний хроматографічний папір.
Рис. 6.16. Газовий хроматограф Agilent 6850. Хроматографія широко застосовується у промисловості, біології, медицині, ветеринарній медицині. Метод є простий і при цьому досліджувані речовини не зазнають хімічних змін, тому що явище адсорбції базується на силах міжмолекулярних взаємодій. Хроматографічний метод дослідження використовується для встановлення амінокислотного складу гідролізатів і первинної структури білків, у вивченні амінокислотного складу плазми та інших біологічних середовищ, при кількісному визначенні вітамінів, гормонів і інших біологічно активних речовин. Черезу високу чутливість хроматографія використовується для виділення різних речовин у чистому вигляді та їх ідентифікації. Тепер хроматографічний аналіз біологічних речовин успішно використовується для діагностики різноманітних захворювань.
КОНТРОЛЬНІ ЗАВДАННЯ 1. Дати визначення адсорбції і назвати її види. 2. Позитивна і негативна адсорбція. 3. Які речовини називають поверхнево-активними? Приклад. 4. Назвати галузі використання поверхнево-активних речовин. 5. В чому суть миючої здатності мила? 6. Дати визначення поняття дифільних молекул. 7. Сформулювати правило Траубе-Дюкло. 8. Вказати різницю між фізичною адсорбцією і хемосорбцією. 9. Чим характеризується адсорбційна рівновага? Як впливає на неї концентрація адсорбтиву і температура? 10. Написати рівняння ізотерми адсорбції Ленгмюра і Фрейндліха. Пояснити значення величин, що входять у рівняння ізотерми. 11. В чому суть гідрофілізації і гідрофобізації поверхні? Яке їх практичне значення? 12. Суть вибіркової адсорбції і її біологічне значення. 13. Поняття про обмінну адсорбцію. 14. Що таке катіоніти і аніоніти? 15. В чому суть хроматографії? Її практичне значення. 16. Класифікація хроматографії за механізмом процесу розділення. 17. В чому суть адсорбційної хроматографії? 18. Де застосовується розділювальна хроматографія? 19. Що лежить в основі йонообмінної хроматографії? 20. В чому суть методу гель-фільтрації (ексклюзивна хроматографія)? 21. Дати загальну характеристику колонкової, тонкошарової, паперової і капілярної хроматографії.
Розділ VIІ КОЛОЇДНОДИСПЕРСНІ СИСТЕМИ §1. Загальна характеристика дисперсних систем. Дисперсними називають системи, які складаються з речовини, подрібненої і розподіленої в іншій речовині. В таких системах подрібнену речовину називають дисперсною фазою, а середовище, в якому ця фаза розподілена – дисперсним середовищем. Так, наприклад, завись глини у воді, емульсія оливи у воді, дим, туман є дисперсними системами. Для характеристики дисперсних систем вживають поняття ступеня дисперсності, тобто ступеня подрібненості речовини дисперсної фази. Ступінь дисперсності D є величиною, оберненою розміру частинок дисперсної фази:
З цього рівняння випливає, що чим більший ступінь дисперсності, тим менший розмір частинок. Важливою ознакою, що характеризує ту чи іншу дисперсну систему, є величина питомої поверхні, яка дорівнює відношенню поверхні частинок до загального об’єму дисперсної фази:
де: Sпит – питома поверхня; S – загальна поверхня частинок, тобто поверхня поділу між фазами; V – об’єм речовини дисперсної фази. Питома поверхня і ступінь дисперсності взаємопов’язані: чим більший ступінь дисперсності, тим менший розмір частинок, тим більша їхня питома поверхня. Всі дисперсні системи за величиною частинок дисперсної фази поділяються на такі групи: грубодисперсні, колоїднодисперсні, молекулярно- та йонодисперсні (істинні розчини). Грубодисперсні системи. Частинки дисперсної фази цих систем мають розміри від 10–3 до 10–5 см. Якщо дисперсним середовищем є вода, розмір молекул якої дорівнює 10–8 см, то у грубодисперсних системах є чітка межа поділу між дисперсною фазою і дисперсним середовищем, тобто вони гетерогенні. До таких систем належать суспензії, емульсії, піни, аерозолі, порошки, пасти. Всі грубодисперсні системи містять частинки дисперсної фази, видимі в звичайний мікроскоп або навіть неозброєним оком. Ці частинки затримує паперовий фільтр. Великі розміри частинок зумовлює нестійкість системи, і з часом дисперсна фаза відокремлюється від дисперсного середовища. Колоїднодисперсні системи (колоїдні розчини) мають розмір частинок від 10–5 до 10–7 см. Вони можуть проходити через пори фільтрувального паперу, але не проходять через рослинні і тваринні мембрани. Колоїдні частинки при наявності у них електричного заряду і сольватно-йонних оболонок не осідають і без зміни умов можуть довго не випадати в осад. Прикладами колоїдних систем є розчини альбуміну, желатини. Колоїдні розчини називають золями. Так, колоїдний розчин дуже подрібненого металічного золота називають золем золота. Залежно від природи дисперсного середовища золі називають гідрозолями, якщо дисперсним середовищем є вода, чи аерозолями, якщо дисперсне середовище – повітря. Так, золь аргентум йодиду у воді – гідрозоль. Молекулярно- та йонодисперсні системи (істинні розчини). В молекулярно- та йонодисперсних системах частинки дисперсної фази і дисперсного середовища мають приблизно однаковий розмір (≈ 10–8 см). У таких системах зникає гетерогенність – системи стають гомогенними, утворюються істинні розчини. До них належать розчини низькомолекулярних речовин-неелектролітів (розчини цукру, спирту та ін.), а також дисперсні системи, в яких речовина є у вигляді окремих гідратованих йонів – водні розчини сильних електролітів (HCl, NaCl, NaOH та ін.). Слабкі ж електроліти (CH3COOH, NH4OH) представлені у водних розчинах одночасно як у вигляді йонів, так і молекул. Зі зміною розмірів частинок від найбільших до найдрібніших і навпаки будуть відповідно змінюватися і властивості дисперсних систем: кінетичні, оптичні, каталітичні та ін. При цьому колоїдні системи займають проміжне положення між грубо-, молекулярно- і йонодисперсними системами (табл. 7.1). Таблиця 7.1. Властивості деяких дисперсних систем. Грубі зависі (суспензії) Колоїдні частини Молекулярно-йонні системи (істинні розчини) Непрозорі Не проходять через паперовий фільтр Не проходять через пергамент Гетерогенні Розсіюють світло в результаті відбиття і заломлення Нестійкі З часом старіють Прозорі (опалесцентні) Фільтруються
Не проходять через пергамент Гетерогенні Дають конус Тіндаля
Відносно стійкі З часом старіють Прозорі Фільтруються
Проходять через пергамент Гомогенні Оптично порожні
Стійкі Не старіють
Колоїдні розчини – гетерогенні системи, і це одна із причин їх нестійкості. Вони володіють значною вільною енергією і, відповідно до другого закону термодинаміки, будуть прямувати до рівноваги, що характеризується розділенням системи на дві фази, які мають мінімальні міжфазові поверхні і, отже, мінімальну поверхневу енергію. Стабілізатори, адсорбуючись на частинках дисперсної фази і знижуючи таким чином величину поверхневої енергії, будуть підвищувати стійкість колоїдних систем. З кінетичних позицій стійкість чи нестійкість гетерогенних систем залежить від величини сил зчеплення і сил відштовхування. Стійкість системи забезпечується вандерваальсовими міжмолекулярними силами. Вони сприяють агрегації частинок. Нестійкі системи характеризуються електричними силами, які запобігають коагуляції частинок. Виникнення електричних сил обумовлене вибірковою адсорбцією на міжфазовій поверхні йонів електроліту. Об’єднанню частинок перешкоджає також утворення на їх поверхні сольватної оболонки, яка складається із молекул дисперсного середовища. Дисперсні системи, і особливо колоїдні, широко розповсюджені у природі. Біологічні рідини тваринних і людських організмів (кров, лімфа, плазма) є колоїдними системами, у яких речовини (наприклад, білки, холестерол, глікоген та ін.) знаходяться в колоїдному стані. Те ж можна сказати і про білки, крохмаль, колоїдні слизі в рослинах. Колоїди різних тканин тваринного і рослинного організмів обумовлюють велику різноманітність їх властивостей (стан драглів, еластичність, набрякання та ін.). Колоїдні речовини можуть зв’язувати велику кількість води (сполучна тканина), а також адсорбувати різноманітні речовини. Адсорбція відіграє важливу роль в обміні речовин, процесах травлення, дії ліків. Найбільшу зацікавленість мають ті дисперсні системи, які найближче стоять до біологічних об’єктів і є колоїдними розчинами. Вони включають високомолекулярну дисперсну фазу і рідке дисперсне середовище (колоїдні розчини білків, поліцукориди та ін.). Загальною характеристикою колоїдних розчинів є властивість їх дисперсної фази взаємодіяти з дисперсним середовищем. У цьому відношенні розрізняють два типи золів. У одних золів частинки мають дуже малу спорідненість з розчинником, погано з ним взаємодіють і утворюють навколо себе тільки тоненьку оболонку із молекул дисперсного середовища. Такі колоїди називають ліофобними (від грецького phobia – ненависть). Якщо дисперсним середовищем є вода, то такі системи називаються гідрофобними. Наприклад, золі металів (заліза, залота), аргентум хлориду та ін. У системах, де диспергована речовина і розчинник мають високу спорідненість, частинки набувають більш об’ємної оболонки із молекул розчинника. Такі системи одержали назву ліофільних (від грецького philia – любов), а у випадку водного дисперсного середовища – гідрофільних колоїдів, як наприклад, розчини білка, крохмалю, агар-агару та ін.
§2. Методи одержання колоїдних розчинів. Однією з основних відмінностей колоїдних розчинів від істинних є їх гетерогенність. Розміри колоїдних частинок є набагато більші (в 10-1000 разів) від розмірів молекул дисперсного середовища і між ними виникає межа розділення. Власне колоїдні розчини є мікрогетерогенні системи, які складаються із дисперсного середовища і дисперсної фази. Такі системи можуть утворюватися за таких умов: 1) розміри диспергованої речовини повинні бути співмірними з розмірами колоїдних частинок; 2) дисперсна фаза повинна б мати погану розчинність; 3) наявність стабілізаторів, які на межі поділу фаз утворюють йонний шар і гідратну оболонку, забезпечуючу зберігання колоїдних частинок у завислому стані. Електричний заряд і гідратна оболонка на поверхні частинок запобігають їх коагуляції і випаданню в осад. Оскільки колоїдні розчини займають проміжне місце за розмірами частинок між грубодисперсними системами та істинними розчинами, для їх одержання можуть бути використані дві групи методів: дроблення – диспергування більших частинок до ступеня дисперсності колоїдних частинок, і агрегація – об’єднання молекул або йонів, що наближуються за розмірами до частинок колоїдних систем. Перший метод одержав назву дисперсний, а другий – конденсаційний. Дисперсні методи Механічне диспергування. Для цього використовуються спеціальні пристрої, так звані шарові та планетарні млини, де матеріал зазнає грубого диспергування. Одержані частинки з розмірами від 10–4 до 10–5 м мелять у вібро- та колоїдних млинах. Ці прилади використовуються для диспергування твердих речовин і рідин у рідкому середовищі при одержанні суспензій та емульсій. Електричне диспергування. Два металевих електроди занурюють кінцями у рідину, потім зближують їх і пропускають струм. Виникає електрична дуга. У її зоні від електродів відриваються частинки, вони надходять у середовище і утворюють золь. Таким способом добувають гідрозолі благородних металів. Колоїдні розчини срібла використовують у ветеринарній медицині як антисептичні лікарські засоби. Ультразвуковий метод ґрунтується на використанні напрямленого ультразвукового поля. Диспергування відбувається внаслідок кавітаційного руйнування, суть якого полягає в тому, що при чергуванні стиснень і розріджень у рідині безперервно утворюються та зникають пустоти. Таке явище зумовлює виникнення механічних руйнівних сил, які спроможні диспергувати як рідини, так і тверді частинки. Під дією ультразвуку відбувається і стерилізація колоїдних розчинів, емульсій, суспензій, тому що руйнуються мікроби і їх спори. Пептизація – це процес переходу речовини із драглеподібного стану в золь під впливом пептизаторів, тобто диспергуючих засобів. Пептизації в основному зазнають пухкі свіжоутворенні осади гідроксидів металів, наприклад Al(OH)3, Fe(OH)3, Zn(OH)2 та ін. Термін “пептизація” (пептинізація) виник за аналогією з процесом розщеплення білка пепсином до пептонів. Пептизація може відбуватися внаслідок видалення із розчину коагулюючих йонів, які викликають укрупнення частинок або адсорбцію пептизатора, що супроводжується утворенням подвійного електричного шару і виникненням сольватної оболонки на колоїдних частинках. В усіх випадках частинки розходяться і внаслідок теплового руху розподіляються по всьому об’єму дисперсного середовища. Пептизаторами є, головним чином, електроліти, які здатні дезагрегувати осади. Прикладом пептизації може бути одержання золя Fe(OH)3 при дії на його осад невеликої кількості розчину FeCl3, взятого як пептизатора. Конденсаційні методи В основі більшості конденсаційних методів одержання колоїдних розчинів лежать різноманітні хімічні реакції: окиснення, відновлення, подвійного обміну, гідролізу та ін. Метод окиснення. В результаті реакції окиснення можна одержати колоїдні розчини, наприклад: 2Н2S+SО2 ® 3S+2Н2О. Атоми сірки конденсуються в колоїдні частинки. Під час реакції утворюється пентатіонова кислота, яка виступає стабілізатором: Н2S5О6 ↔ 2Н++S5О62–. Метод відновлення базується на введенні в розчин солей металів будь-якого відновника: спирту, таніну, формаліну, гідразину, гідроген пероксиду. Втрачаючи свій заряд, йони металу перетворюються на частинки металу, які між собою агрегують і у присутності надлишку солі металу (стабілізатора) досягають колоїдних розмірів. Золь золота можна одержати відновленням калій аурату формальдегідом:
Cтабілізатором виступає калій аурат: КАuО2 ↔ К++АuО2–. Часто процес відновлення призводить до утворення частинок, розмір яких перевищує розмір колоїдних частинок. Щоб запобігти цьому збільшенню розмірів частинок, процес відновлення проводять у присутності розчинів високомолекулярних сполук (ВМС). Їх молекули адсорбуються на поверхні колоїдних частинок, утворюючи захисну плівку, яка перешкоджає злипанню. Так, при одержанні лікарських препаратів – коларголу та протарголу, які є колоїдним розчином срібла, використовують солі лізальбінової та протальбінової кислот як захисний засіб від злипання колоїдних частинок. Метод подвійного обміну дає змогу одержати золі важкорозчинних сполук. Наприклад, при змішуванні розведених розчинів BaCl2 та K2SO4 за умови, що один з реагентів є у надлишку, барій сульфат не випадає в осад, а утворює колоїдний розчин:
BaCl2+K2SO4 → 2KCl+BaSO4 надлишок важкорозчинна сполука
Метод гідролізу. Гідроліз широко використовується при одержанні золів із солей, якщо в результаті реакції гідролізу утворюється високорозчинна речовина. Так, золь гідроксиду заліза одержують реакцією: FeCl3+3H2O = Fe(OH)3+3HCl. Проміжним продуктом гідролізу є ферум (ІІІ) хлороксид: FeCl3+H2O = FeOCl+2HCl; FeOCl ↔ FeO++Cl–. Утворені йони забезпечують йоногенний шар навколо частинок Fe(OH)3 і утримують їх у зависі.
Рис. 7.1. Схема одержання коло-їдних розчинів металів. Електричний метод одержання колоїдних розчинів запропонував Бредінг у 1898 році. Метод використовується в основному для приготування гідрозолів благородних металів. Виникає електрична дуга між електродами диспергуючого металу (золото, срібло та ін.). Під дією високої температури відбувається випаровування металу електродів у дисперсному водному середовищі. Випари металу конденсуються у колоїдні частинки, утворюючи відповідний золь (рис. 7.1).
§3. Методи очищення колоїдних розчинів. Для одержання стійких колоїдних розчинів та вивчення їх властивостей необхідним є видалення із золів різних домішок і в першу чергу надлишку електролітів, які утворюються при одержанні колоїдних розчинів. Для цього використовуються діаліз і ультрафільтрація. Діаліз – це метод очищення колоїдних розчинів від домішок, що можуть проникати через рослинні, тваринні і штучні мембрани. Прилади, які використовуються для діалізу, називаються діалізаторами.
Рис. 7.2. Схема діалізатора. В основі будови і дії діалізаторів (рис. 7.2) є посудина, заповнена проточною водою. У посудину на певну глибину занурюють другу посудину, дно якої виготовлене з мембрани. За допомогою діалізу поступово відбувається видалення речовин, які легко проникають крізь мембрану, наприклад, електролітів та інших кристалоїдів. Таке очищення колоїдних розчинів відбувається дуже повільно, і тому для прискорення діалізу запропоновано використовувати електричний струм.
Рис. 7.3. Схема електродіа-лізатора. Електродіаліз. Прискорений процес діалізу – електродіаліз. Прилад для електродіалізу є посудина, розділену двома мембранами М на три частини (рис. 7.3). У середню частину А вносять колоїдний розчин, а зовнішні В і С, у яких вмонтовані електроди, заповнюють розчинником – проточною водою. При пропусканні електричного струму відбувається напрямлений рух катіонів і аніонів до відповідних електродів. Катіони і аніони проникають через мембрану, а колоїдні частинки – ні. Таким чином вміст йонів у колоїдному розчині постійно зменшується, тобто відбувається очищення колоїдного розчину. Електродіаліз використовується у медицині і ветеринарії для очищення біопрепараторів, білкових речовин, гормонів, антибіотиків та ін. За допомогою електродіалізу хворому через шкіру вводять деякі лікарські речовини (йонофорез). Компенсаційний діаліз і вівідіаліз Міхаелісом і Роном, для дослідження біологічних рідин, запропонований метод, який дозволяє визначити концентрацію деяких низькомолекулярних сполук, що перебувають у вільному стані в колоїдних розчинах. Суть компенсаційного діалізу полягає в тому, що рідина в діалізаторі омивається не чистим розчинником, а розчинами визначуваної речовини різних концентрацій. Так, наприклад, незв’язаний з білками цукор сироватки крові визначається шляхом діалізу сироватки проти ізотонічного сольового розчину, у який вносять різні кількості цукру. Концентрація цукру в сольовому розчині при діалізі не буде змінюватися лише у тому випадку, коли вона рівна концентрації вільного цукру у сироватці. Цей метод дає змогу визначити істинні концентрації речовин у колоїдних розчинах. Приблизно на цьому ж принципі розроблений метод вівідіалізу (vivus – життя), який виконується у живому організмі тварин і людей. У кінці перерізаної кровоносної судини вмонтовують скляні канюлі, розгалужені частини яких сполучаються між собою колодієвими трубками і вся система занурюється у посудину з фізіологічним розчином NaCl або водою. Так було виявлено вільні амінокислоти крові. На принципі компенсаційної вівідифузії сконструйований апарат, що одержав назву “штучної нирки”. За допомогою цього пристрою можна видаляти з крові продукти обміну і, отже, тимчасово замінювати функцію нирки. Робота “штучної нирки” базується на принципі діалізу речовин через напівпроникну мембрану. Діалізуючий розчин містить усі низькомолекулярні речовини, які необхідно зберегти у складі крові, причому в однакових з кров’ю концентраціях. В ході діалізу із крові видаляються непотрібні і шкідливі речовини (сульфати, фосфати, сечова кислота, сечовина та ін.). Показанням для використання “штучної нирки” є гостра ниркова недостатність, наприклад, при отруєнні сулемою, сульфаніламідними препаратами, при уремії після переливання крові, при важких опіках і т. п. Ультрафільтрація – метод очищення колоїдного розчину фільтруванням його крізь напівпроникну мембрану, яка пропускає дисперсне середовище разом з низькомолекулярними домішками, але затримує частинки дисперсної фази. Для прискорення ультрафільтрації цей процес проводять при різниці тисків по обидва боки мембрани. Ультрафільтрація дає змогу швидше відокремити від колоїдного розчину електроліти та інші низькомолекулярні органічні сполуки, ніж це відбувається при діалізі. Ультрафільтрація широко використовується у промисловості і науково-дослідній роботі, в медицині та ветеринарії (очищення від йонних і нейонних домішок води, рідкого палива, оливи, розчинників, білків, нуклеїнових кислот, ферментів, вітамінів та ін.). Вона використовується при аналізі забруднень довкілля промисловими відходами, в мікробіології і вірусології. Ультрафільтрація може бути поєднана з електродіалізом – електроультрафільтрація. В діалізаторі монтується ультрафільтр з електродами. Електроультрафільтрацію застосовують для очищення та розділення білків, а в останні роки в лікарнях для обробки крові. Електродекантація застосовується для підвищення концентрації золю або розчину ВМС. Для цього в середню частину звичайного електродіалізатора вноситься колоїдний розчин і пропускається електричний струм і вода. Заряджені колоїдні частинки рухаються до відповідного електрода, в результаті чого зростає їх концентрація. Концентрований розчин відводиться і збирається у окремий посуд. Цей спосіб концентрування вперше був використаний для одержання деяких вірусів у чистому вигляді, наприклад, віруса поліомієліту.
§4. Будова колоїдних частинок. Окрема колоїдна частинка називається міцелою (від новолат. micella – крупина, дрібка). Вона складається із внутрішньої і зовнішньої частин. Внутрішня частина міцели називається ядром, яке може мати аморфний або кристалічний стан. Зовнішня частина складається з адсорбційного і дифузного шарів. Адсорбційний шар щільно прилягає до ядра. Він утворюється адсорбованими молекулами або йонами стабілізуючої речовини. Якщо такий шар має йонну природу, то в основі шару йони одного знаку і деяка кількість йонів протилежного знаку (протийони). Дифузний шар менш щільний. Він запобігає зближенню часток у процесі броунівського руху. Число позитивних та негативних зарядів у міцелі однакове, і вона в цілому електронейтральна. Ядро міцели – це асоціат сотень і тисяч молекул. При утворенні ядра молекули об’єднуються так, що гідрофобні ділянки утворюють в ньому внутрішні, а гідрофільні – зовнішні зони. Якщо дисперсним середовищем є органічні речовини, то радикали (ліофобні ділянки) повернуті назовні. Будову колоїдних частинок краще розглядати за їх утворенням. Будова міцели AgJ. Утворення міцели AgJ відбувається у процесі хімічної реакції: AgNO3+KJ → AgJ↓+KNO3; Ag++NO3–+K++J– → AgJ↓+K++NO3–. Якщо калій йодиду і аргентум нітрату взято в еквівалентних кількостях, частинки AgJ зростають, досягаючи значних величин, що перевищують розміри колоїдних частинок, а потім швидко випадають в осад. Якщо ж реакцію проводити з дуже розбавленими розчинами при невеликому надлишку одного з реагентів, то осад не випадає, а утворюється колоїдний розчин аргентум йодиду. Ядро колоїдної частинки, що має кристалічну структуру, нерозчинне у дисперсному середовищі. Воно становить основну масу міцели і побудоване з нейтральних молекул або атомів. У цьому прикладі ядро – найдрібніший кристалик аргентум йодиду, що складається з великої кількості молекул AgJ (mAgJ). Ядро колоїдного ступеня дисперсності є носієм вільної поверхневої енергії, тому на його поверхні проходить адсорбційний процес. Згідно з правилом Пєскова-Фаянса, на поверхні ядра міцели адсорбуються йони, які є в складі ядра міцели, тобто адсорбуються йони срібла або йони йоду – ті, які є в надлишку. Якщо добувати колоїдний розчин при надлишку калій йодиду, то адсорбуватимуться йони йоду, бо вони є в надлишку. Йони йоду добудовують кристалічну решітку ядра, міцно входять в його структуру, утворюючи адсорбційний шар, і надають ядру негативного заряду: m[AgJ]nJ–. Ці йони, що адсорбуються на поверхні ядра і надають йому відповідного заряду, називаються потенціаловизначальними йонами. У розчинні є також і йони, протилежні за знаком потенціаловизначальним, їх називають протийонами. У цьому прикладі – це катіони К+, що електростатично притягуються потенціаловизначальними йонами адсорбційного шару. Ядро з адсорбційним шаром називають часточкою, або гранулою:
{m[AgJ]·nJ–·(n–x)K+}x–. гранула В адсорбційному шарі гранули переважають потенціаловизначальні йони. Кількість їх можна позначити nJ–, а кількість протийонів – (n–x). Решта протийонів утворює дифузний шар йонів. Ядро з адсорбційним і дифузним шарами називають міцелою: {m[AgJ]·nJ–·(n–x)K+}x–·хК+. міцела Якщо добувати сіль аргентум йодиду при надлишку аргентум нітрату, тобто при надлишку Ag+, то колоїдна частинка завдяки адсорбції йонів Ag+ на поверхні ядра дістане позитивний заряд. На рис. 7.4 схематично зображено міцели золю аргентум йодиду, добутого при надлишку Ag+ (а) і при надлишку калій йодиду (б): а) гранула (позитивна)
б) гранула (негативна)
Числа m, n, x залежно від умов приготування колоїдних розчинів можуть змінюватися в широких межах, тобто міцела не має чітко визначеного складу. В цілому вона є електронейтральна колоїдна частинка, здатна самостійно існувати. Вона визначає всі основні властивості колоїдної системи. Коли пропускати постійний електричний струм крізь колоїдний розчин, до електродів рухатимуться не міцели, що є електронейтральними, а тільки гранули. За напрямленістю переміщення гранул в електричному полі вдалося довести, що частинки золів мають однойменний (негативний або позитивний) заряд.
Рис. 7.4. Будова міцел золю срібла йодиду, добутого при надлишку AgNO3 (a) і при надлишку КJ (б). Наявність однойменного заряду в усіх частинках золю – важливий фактор його стійкості. Заряд перешкоджає злипанню і збільшенню колоїдних частинок, тобто коагуляції. Стійкість колоїдних частинок пояснюється тим, що на поверхні ядер адсорбується певний вид потенціаловизначальних йонів. Ті електроліти, йони яких є потенціаловизначальними, слід вважати стабілізаторами, а йони, що адсорбуються поверхнею ядер – стабілізуючими йонами. При цьому на ядрі адсорбуються ті йони – стабілізатори, які містять у собі елементи, спільні з ядром. Наприклад, золь заліза гідроксиду (ІІІ): m[Fe(OH)3] ядро колоїду nFeCl3 → nFe3++3nCl– йонний стабілізатор {m[Fe(OH)3·nFe3+·3(n–x)Cl–}3x+·3xCl– міцела
§5. Молекулярно-кінетичні властивості колоїдних систем. Цілим рядом властивостей колоїдні розчини відрізняються як від істинних розчинів, так і від грубодисперсних систем. І перш за все за молекулярно-кінетичними властивостями, які залежать від величини, форми та інтенсивності руху колоїдних частинок. Броунівський рух. У 1827 році ботанік із Великої Британії Роберт Броун, вивчаючи під мікроскопом квітковий пилок у краплі води, виявив, що частинки пилку безперервно і безладно переміщуються. Цей хаотичний рух частинок одержав назву броунівського руху. Численними дослідженнями встановлено, що броунівський рух залежить від температури, в’язкості дисперсного середовища і розмірів частинок. Згідно з теорією броунівського руху, розробленою А.Айнштайном (1900), будь-яка рідина чи газ складається з молекул, які перебувають у русі. Імпульси руху окремих молекул різні за величиною і напрямком. Якщо в таке середовище потрапляє частинка, то вона зазнає впливу з боку молекул середовища. Частинки набувають хаотичного руху, змінюючи напрям і величину швидкості приблизно 1014 разів на секунду. При малих розмірах частинок число ударів неоднакове з різних боків, і тому вони рухаються у різних напрямках по складній траекторії (рис. 7.5).
Рис. 7.5. Броунівський рух частинок. Зі збільшенням розмірів і маси частинок ймовірність взаємної компенсації ударів зростає, внаслідок цього частинки розміром 4-5 мк здійснюють лише невеликий коливальний рух біля якогось центра рівноваги. При діаметрі частинок більше 5 мк броунівський рух практично припиняється. Інтенсивність броунівського руху залежить від розмірів частинок, внутрішнього тертя, в’язкості середовища, абсолютної температури, коефіцієнта дифузії та ін. Залежність зміщення частинок
Коефіцієнт дифузії для сферичних частинок, які мають значно більший розмір, ніж молекули розчинника, виражається рівнянням:
Це рівняння дозволяє на основі виміру D визначати радіус колоїдних частинок, форму, а також величину молекул. Ввівши у рівняння (7.3) значення D, для середнього зміщення частинок за одиницю часу одержимо:
де: Вчення про броунівський рух показало, що колоїдні розчини можна розглядати як істинні розчини, у яких розчинена речовина міститься у вигляді частинок, що володіють властивостями броунівського руху. Дифузія і осмотичний тиск. Під впливом теплового і броунівського руху відбувається процес вирівнювання концентрації частинок по всьому об’єму розчину. Процес дифузії відбувається не тільки в молекулярних, але й у колоїднодисперсних розчинах. Айнштейн встановив залежність між коефіцієнтом дифузії і радіусом дифундуючих частинок (7.4). Колоїдні частинки обумовлють також і деякий осмотичний тиск. Внаслідок великих розмірів частинок і малих концентрацій їх осмотичний тиск дуже малий. Наприклад, 1% розчин цукрози (істинний розчин) має осмотичний тиск 0,0725 МПа, тоді як розчин золю золота тієї ж концентрації і за тих же умов має осмотичний тиск 0,000045 МПа. Крім того, частина величини осмотичного тиску в колоїдних розчинах припадає на домішки електролітів. Осмотичний тиск колоїдних розчинів вимірюється спеціальними осмометрами. Колоїдні розчини володіють малим зниженням пружності пари, низькими величинами зниженням температури замерзання і підвищенням температури кипіння. Наприклад, температура замерзання золю золота при концентрації 1 г/л і розмірі частинок 4 нм менша, ніж чистого розчинника (води) на 0,000004ºС. Тому методи кріоскопічного та ебуліоскопічного визначення величини осмотичного тиску для них не придатні. Залежність осмотичного тиску розчинів від величини дисперсності їх частинок характеризується відповідними рівняннями. Так, якщо два колоїдних розчини мають одинакову дисперсну фазу і вагову концентрацію С, але різні розміри частинок, то вагова кількість диспергованої речовини в одиниці об’єму дорівнює: С = ¾3πr3dn, (7.6) де: d – густина розчину; n – число частинок в одиниці об’єму; r – радіус частинок. У випадку, коли у двох колоїдних розчинах однакове дисперсне середовище, при однаковій температурі їх осмотичний тиск можна виразити рівнянням:
Отож, осмотичний тиск колоїдних розчинів обернено пропорційний кубу радіуса їх частинок і прямо пропорційний кубу їх ступеня дисперсності. Седиментація (від лат. sedimentum – осідання) – осідання частинок дисперсної фази в рідині чи газі під впливом гравітаційного поля або відцентрових сил. У колоїдних розчинах седиментація настає лише при укрупненні частинок у результаті перекристалізації або їх коагуляції. Частинки дисперсної фази, що знаходяться в газоподібному стані або рідкому дисперсному середовищі, зазнають дії двох взаємопротилежних сил: сили тяжіння намагаються сконцентрувати частинки у нижніх шарах і сили дифузії намагаються перемістити дисперсну фазу аерозолів, колоїдних розчинів із зон більших концентрацій в зони менших концентрацій. Здатність дисперсної системи зберігати рівномірний розподіл частинок по всьому об’єму називається седиментаційною або кінетичною стійкістю. Високодисперсні системи (істинні розчини) володіють значною кінетичною стійкістю і, навпаки, грубодисперсні системи є кінетично нестійкі, оскільки їх частинки практично не можуть здійснювати тепловий рух. Колоїдні розчини займають між ними проміжне положення. Швидкість седиментації розраховують за рівнянням:
де: 218 ≈ 2/9 g ; g – прискорення сили тяжіння; υ – швидкість осідання, см/сек; r – радіус частинок; η – в’язкість; d1 – густина диспергованої речовини; d2 – густина розчинника. Залежність величини радіуса і швидкості осідання наведені в табл. 7.2. Таблиця 7.2. Швидкість осідання у воді частинок різної величини. Радіус кулястої частинки, мкм Швидкість осідання см/сек Час проходження частинкою шляху в 1 см 0,1 0,01 0,001 1.706·10–1 1.706·10–3 1.706·10–5 1.706·10–7 1.706·10–9 5,86 сек 9,8 хв 16 год 68 діб 19 років
Седиментація і седиментаційний аналіз застосовується у промисловості, клінічній практиці, науковій роботі. Так, лікувальна дія багатьох ліків (паст, порошків і зависів) залежить від ступеня дисперсності. Якісна одиниця функціонального стану еритроцитів (РОЕ) дозволяє лікарю роботи висновки про стан захисних сил організму людини і тварин. Ультрацентрифугування. Осідання частинок колоїдів під дією сил тяжіння відбувається поволі. А.Думанський у 1912 році запропонував для визначення константи седиментації використовувати центрифугування. Через одинадцять років Т.Сведбергом вперше була сконструйована спеціальна центрифуга – ультрацентрифуга. Сучасні ультрацентрифуги розвивають швидкість понад 60 000 об/хв. Потужність таких центрифуг характеризується величиною g, яка показує, у скільки разів перевищує прискорення сил тяжіння, наприклад, 140 000 g, 150 000 g і т. д. Розрізняють аналітичні і препаративні ультрацентрифуги. Аналітичні ультрицентрифуги мають оптичну систему, яка дозволяє реєструвати на фотострічках результати ультрацентрифугування. В аналітичних центрифугах проводиться визначення констант седиментації частинок (константа седиментації частинок виражається у Сведбергах S = 1·103 см/сек·дін), що дозволяє обчислювати їх відносні молекулярні маси.
Рис. 7.6. Препаративна ультрацентрифуга Hitachi 85P-72. Препаративні ультрацентрифуги (рис. 7.6) призначені для виділення із розчинів окремих фракцій. Фракції можуть бути однорідні тільки за одним показником – за швидкістю седиментації, а за іншими властивостями можуть значно різнитися. Наприклад, γ-глобуліни крові при центрифугуванні розділяються на дві фракції з константами седиментації 7S і 19S. Білки ж, які утворюють ці фракції, різняться між собою рядом властивостей (імулогічним, електрофоретичним і т. п.). За допомогою імунохімічних методів аналізу кожну із цих фракцій можна розділити на нові фракції. Так, виявилося, що до 7S-глобулінів належать γА- і γG-глобуліни. Ці фракції у свою чергу складаються із семи білків, які мають деякі фізико-хімічні відмінності. Ультрацентрифуги широко використовуються при вивченні білків, нуклеїнових кислот, вірусів, різних клітинних структур. §6. Оптичні властивості колоїдних систем. Оптичні властивості колоїдних систем зумовлюються їх дисперсністю, гетерогенністю, розсіюванням світла. При проходженні світла крізь дисперсну систему спостерігаються явища заломлення, відбиття, розсіювання і поглинання. У грубодисперсних системах розмір частинок перевищує довжину хвилі видимої частини спектра. Це зумовлює відбиття світла від поверхні частинок. У золях колоїдних розчинів розмір частинок співмірний із довжиною хвилі видимого світла. Спостерігається не відбиття, а розсіювання світла. Розсіювання світла характерне для будь-якого середовища. Найінтенсивніше світло розсіюється у колоїдних або ультрамікрогетерогенних системах з розміром частинок від 1 до 100 нм. При цьому лінійні розміри частинок менші за довжину падаючого світла і це зумовлює явище дифракції: світлові хвилі огинають частинки і змінюють свій початковий напрям. Дифракція є причиною матового світіння – опалесценсії.
Рис. 7.7. Розсіювання світла колоїдними час-тинками (конус Тіндаля). Дифракційне розсіювання світла вперше було помічене М.Ломоносовим, а пізніше – М.Фарадеєм і його учнем Дж.Тіндалем (1869) при пропусканні світлового променя через колоїдний розчин золота. В темному приміщенні при боковому спостереженні проходження світлового променя крізь скляну посудину з колоїдним розчином Тіндаль виявив світловий конус, який виникав внаслідок розсіювання світла колоїдними частинками. Це явище одержало назву конуса Тіндаля, за яким легко відрізнити колоїдні розчини від істинних розчинів (рис. 7.7). Аналогічне явище – світіння пилинок у повітрі – можна спостерігати при проникненні тонкого світлового пучка у темне приміщення або ліхтарного променя на темному фоні неба. Теорію світлорозсіювання (опаласценсії) розвинув Д.Релей (1871). Загальна кількість світлової енергії, що розсіюється одиницею об’єму дисперсної системи, дорівнює:
якщо
то тоді
де: I0 – інтенсивність падаючого світла; n1 і n0 – показники заломлення дисперсної фази і дисперсного середовища; ν – число частинок в одиниці об’єму; V – об’єм однієї частинки; λ – довжина хвилі падаючого світла; К – константа, яка залежить від показників заломлення дисперсного середовища і дисперсної фази. З наведеного рівняння Релея випливає, що розсіювання світла тим більше, чим більше показник заломлення дисперсної фази n1 різниться від показника заломлення дисперсного середовища n0. При n1 = n0 світло не розсіюється. Закон Релея дозволяє судити про концентрацію частинок в зависях. На цьому принципі сконструйовані нефелометри, за допомогою яких визначають інтенсивність світла, що розсіюється дисперсною системою. В основі роботи нефелометра лежить явище опалесценсії. За допомогою нефелометрів визначають концентрацію і середню величину колоїдних частинок. З рівняння (7.10) добуток νV є загальним об’ємом дисперсної фази в одиниці об’єму розчину. Масова концентрація дисперсної фази: c = ρνV, (7.11) де ρ – густина дисперсної фази. Звідси:
і тоді
За відповідних значень дисперсної фази і дисперсного середовища при використанні певного джерела освітлення величини К, ρ і λ є сталими. Тоді рівняння (7.12) набуде вигляду: Ip = КсV. (7.13) Це рівняння є основою для розрахунків при нефелометричних вимірюваннях концентрації білка в сечі тварин і людини. Метод базується на властивості білка утворювати каламутність з сульфасаліциловою кислотою. Інтенсивність каламутності пропорційна концентрації білка. Забарвлення золів. Рівняння Релея виражає залежність інтенсивності розсіювання світла від об’єму частинок і довжини хвилі падаючого світла. У першому випадку інтенсивність розсіяного світла прямопропорційна довжині світлової хвилі у четвертому ступені. Отже, якщо світлове джерело (наприклад, промінь видимого спектра) складається з хвиль різної довжини, то найкоротші (голубі), попадаючи на колоїдні частинки, будуть розсіюватися сильніше від інших. Тому цілий ряд колоїдних систем: гідрозолі каніфолі, сульфуру, срібла хлориду, дим у відбитому світлі (тобто при розгляді під кутом до напрямку падаючих променів), – будуть мати голубувате забарвлення. Червонувате або червонувато-жовте забарвлення золів у падаючому світлі обумовлене тим, що короткохвильова частина спектра (синьо-фіолетова) розсіюється сильніше, ніж довгохвильова частина спектра (жовто-червона). Залежність інтенсивності розсіювання світла від довжини хвилі використовується для світлової сигналізації: при світломаскуванні використовуються сині лампи, тому що синє світло, проходячи крізь шар атмосфери, що містить велику кількість різних частинок пилу, диму, туману, сильно розсіюється і тому не буде видимим, наприклад, з літака. І навпаки, для сигналізації про небезпеку використовують червоні лампи. Ультрамікроскопія і електронна мікроскопія. В основі методу ультрамікроскопії лежить дослідження об’єкта у звичайному мікроскопі з високою роздільною здатністю при бічному освітленні. Сильне джерело світла і система лінз створює яскравий променевий пучок, який проходить крізь колоїдний розчин, не потрапляючи в око спостерігача. Завдяки світлорозсіюванню стають видимими окремі колоїдні частини у вигляді світлих точок на темному фоні. Ультрамікроскоп дає змогу бачити частинки у вигляді світлої точки з лінійними розмірами від 10 до 300 нм. Ним можна встановити швидкість руху окремих частинок, концентрацію їх в об’ємі, спостерігати коагуляцію у вигляді злиття двох світлих частинок, робити висновки про їх форму. Якщо прийняти для частинок сферичну або кубічну форму, то можна розрахувати розміри частинок – радіус r або довжину ребра l. Для цього слід знати масову концентрацію дисперсної фази (7.11). Розрахунок ведуть за формулами:
або
Електронна мікроскопія – один з сучасних досконалих методів визначення розмірів і форм частинок. Основною відмінністю електронного мікроскопа від оптичного є використання потоку електронів замість світлового променя, а роль оптичних лінз відіграють електромагнітні лінзи. Джерелом електронів служить вольфрамова дротина, яка при сильному нагріванні випромінює потік електронів. Електрони, пролітаючи через електромагнітну систему, попадають на фотопластинку чи екран і дають збільшене зображення предмета (рис. 7.8). За допомогою електронного мікроскопа досконало вивчені віруси. Цей прилад дозволяє бачити мікроби, фаги, структурні елементи клітини, деякі великі молекули.
Рис. 7.8. Схема будови електронного мікроскопа. §7. Електрокінетичні явища. Електрокінетичні явища ґрунтуються на взаємозв’яку між електричними і кінетичними властивостями дисперсних систем. На поверхні ядер колоїдних частинок утворюється подвійний (адсорбційний і дифузний) електричний шар. Адсорбційний шар містить у собі потенціаловизначальні йони і частину протийонів, решта протийонів становить дифузний шар. Коли рідина рухається відносно твердої поверхні, на межі адсорбційної і дифузної частин подвійного шару виникає різниця потенціалів, яку називають електролітичним потенціалом або дзета-потенціалом ξ. Величина цього потенціалу залежить від товщини дифузної частини подвійного шару. Чим товщий дифузний шар, тим більший електрокінетичний потенціал, тим більші сили електростатичного відштовхування, що діють між колоїдними часточками, тим вища стійкість колоїдної системи. Коли йони дифузного шару переходять в адсорбційний, товщина дифузного шару зменшується, знижується електрокінетичний потенціал і стійкість колоїдної системи. За певних умов електрокінетичний потенціал дорівнює нулю – колоїдна система перебуває в ізоелектричному стані. При цьому дифузний шар зникає, що сприяє зближенню колоїдних частинок до таких віддалей, коли сили міжмолекулярного зчеплення стають більшими за електростатичне відштовхування і часточки починають злипатися в більші агрегати, тобто колоїдна система стає агрегативно нестійкою. Слід мати на увазі, що коагуляція настає не в ізоелектричному стані, а дещо раніше. Явища відносного руху фаз уздовж поверхні поділу, які спричинюють до виникнення електричного потенціалу, називають електрокінетичними. Це електроосмос, електрофорез, потенціал перебігу та потенціал седиментації. На основі цих електрокінетичних явищ можна виміряти величину ξ-потенціалу. Електроосмос. Електрокінетичні явища у колоїдних розчинах були відкриті Ф.Рейссом (1808) при дослідженні електролізу води. Вивчаючи причини підвищення рівня води в циліндрі з негативно зарядженим електродом, Ф.Рейсс провів такий дослід: перегородив нижню частину U-подібної трубки діафрагмою з кварцового піску і заповнив трубку водою. Під дією зовнішнього електичного поля рідина переміщувалася до негативно зарядженого електрода. Рух рідини відбувався доти, доки не встановиться певна різниця рівнів у U-подібній трубці (рівновага з гідростатичним тиском). Оскільки без діафрагми переміщення рідини не відбувається , то Ф.Рейсс зробив висновок: при контакті з частинками кварцового піску рідина заряджається. Згодом це явище спрямованого переміщення дисперсного середовища відносно нерухомої дисперсної фази в постійному електричному полі одержало назву електроосмосу. Напрям переміщення рідини вказує на знак ξ-потенціалу, тобто на знак заряду твердої поверхні на межі з рідиною. Вимірювання швидкості руху рідини дає можливість розрахувати стрибок потенціалу на поверхні “ковзання”. Для сталої лінійної швидкості рідини відносно мембрани можна одержати рівняння на підставі уявлення про подвійний електричний шар як плоский конденсатор. Рівняння отримало назву його авторів Г.Гельмгольца та М.Смолуховського і має вигляд:
або
де: η – в’язкість рідини; υ0 – лінійна швидкість течії рідини; Н – напруга зовнішнього електричного поля (градієнт потенціалу), Величину, яка рівна відношенню швидкості руху дисперсного середовища до одиниці напруги електричного поля, називають електроосмотичною швидкістю:
Підставивши це значення у (7.16), одержують:
У вираз для визначення ξ – потенціалу входить лінійна швидкість течії рідини υ0, яку зручніше замінити на об’ємну швидкість. Відношення лінійної швидкості руху рідини до напруги електричного поля (градієнт потенціалу) можна перетворити, користуючись законом Ома, у співвідношення:
де: υx – об’ємна швидкість течії рідини; І – сила струму. Якщо підставити значення υ0 із (7.18) у (7.16), одержимо формулу для розрахунку ξ – потенціалу:
Усі величини, що входять у це рівняння, можна виміряти експериментально.
Рис. 7.9. Зональний електрофорез: 1 – фільтрувальний папір, просочений бу-ферним розчином, він з’єднує з елект-родними камерами носій 2, на якому переміщуються білкові зони (заштри-ховані). Електрофорез (рис. 7.9). Проводячи інший експеримент з електролізу води, Ф.Рейсс у шматочок сирої глини помістив дві скляні трубки (рис. 7.10), на дно яких насипав чистого піску, трубки заповнив водою і вмонтував у них електроди. При пропусканні електричного струму через деякий час в анодній трубці з’являлися частинки глини (каламуть). Явище спрямованого переміщенням частинок дисперсної фази в електричному полі називають електрофорезом.
Рис. 7.10. Схема досліду Рей-сса: 1 – вода, 2 – пісок, 3- сус-пензія глини, 4 – сира глина, 5 – перенесені часточки глини, 6 – переміщення води. Його можна спостерігати лише в седиментаційно стійких дисперсних системах. Рухливість частинок в електричному полі зумовлена тим, що при накладанні на таку систему зовнішньої різниці потенціалів відбувається розрив подвійного електричного шару по площині “ковзання”. Внаслідок цього частинка одержує певний заряд і переміщується до протилежно зарядженого електрода. При цьому протийони дифузного шару рухаються у протилежному напрямку. Швидкість руху частинок дисперсної фази пропорційна величині їх ξ-потенціалу. Рівняння (7.16) справедливе і для електрофорезу. Але лінійна швидкість руху частинок υ0 не може бути їх характеристикою, бо вона змінюється пропорційно напрузі зовнішнього електричного поля Н. У зв’язку з цим введено поняття електрофоретичної рухливості υеф. Остання дорівнює швидкості руху частинок при одиничному градієнті потенціалу (Н = 1):
звідки:
ξ = Вυеф. (7.21) Лінійна швидкість υ0 – це відношення лінійного зміщення межі золю S до одиниці часу τ
При розрахунках ξ-потенціалу частинок, що знаходяться у розведених водних розчинах при 20ºС, можна використати співвідношення: ξ = 2,1·106 υеф – для часточок сферичної форми; ξ = 1,4·106 υеф – для часточок циліндричної форми, де υеф вимірюється в м2/(сВ), а ξ-потенціал в В або mВ. Значення ξ-потенціалу колоїдних часточок перебуває в межах 1,5-75 mВ, а експериментальні значення електрофоретичної рухливості частинок золів має значення порядку υеф = 0,4-0,8·10–8 м2/(сВ); для еритроцитів корів υеф = –1,0-1,7·10–8 м2/(сВ). Електрофорез застосовується для осадження золів, емульсій і суспензій фарб на металічних поверхнях. За його допомогою розділяють білки на фракції і підфракції. Електрофоретичний аналіз біологічних рідин використовується у клінічній практиці для діагностики багатьох захворювань (рис. 7.11). Електрофорез застосовується у фізіотерапії для введення через шкіру чи слизові оболонки ліків хворим (йонофорез). Електрофорез клітин знайшов застосування при оцінці клітинного імунітету в онкологічних хворих.
Рис. 7.11. Ізоензимні спектри лактатдегідрогенази (ЛДГ) си-роватки крові: а –здорової лю-дини; б – хворого на інфаркт міокарда; в – при гепатиті В. Електрофорез клітинних частинок є особливо цінним для гематології та імунології. Всі клітини крові хребетних мають від’ємний заряд, однак кожний тип клітин характеризується певним значенням електрокінетичного потенціалу. За електрофоретичною рухливістю клітини крові можна розмістити у такій послідовності: гранулоцити –0,6·10–12 м2/(сВ); лімфоцити –0,8·10–12 м2/(сВ); еритроцити –1,0·10–8 м2/(сВ). Потенціали перебігу і седиментації. Явище, обернене до електроосмосу, є потенціалом течії або перебігу. Розділення йонів різних знаків та перенесення зарядів по капіляру призводить до появи на кінцях капіляра різниці потенціалів – потенціалу перебігу. Останній є джерелом виникнення біопотенціалів. При протіканні крові по капілярах кровоносної системи виникає потенціал перебігу, який зображується Q-зубцем на кардіограмі. Потенціал перебігу U пропорційний перепаду тиску ∆p. Рівняння Гельмгольца-Смолуховського (7.16) для розрахунків ξ-потенціалу за врахуванням потенціалу перебігу матиме вигляд:
де: η – в’язкість; χ – питома електропровідність; U – потенціал перебігу; р – тиск; ε – діелектрична проникність; ε0 = 8,854·10–12 Потенціал седиментації виникає при роботі центрифуг. Явище потенціалів перебігу і седиментації спостерігається при зсіданні суспензій та емульсій, при розділенні фаз.
§8. Стійкість і коагуляція колоїдних систем. Види стійкості гідрофобних золів. Під стійкістю дисперсної системи розуміють постійність в часі її стану і основних властивостей: дисперсності, рівномірного розподілу частинок дисперсної фази в об’ємі дисперсного середовища і характеру взаємодії між частинками. Питання стійкості дисперсних систем займають центральне місце в колоїдній хімії, тому що основний клас колоїдних систем – ліофобні колоїди – термодинамічно нестабільні, тобто схильні до коагуляції. Коагуляція – це процес злипання (або злиття) частинок дисперсної фази при втраті системою агрегативної стійкості. Надання системам стійкості вимагає спеціальних методів стабілізації. Тільки за таких умов можна одержувати і використовувати багато цінних матеріалів, продуктів, лікарських препаратів, аерозольних засобів та ін. М.Пєсков (1920) ввів поняття про види стійкості дисперсних систем: седиментаційний (кінетичний) і агрегативний. Седиментаційна стійкість дозволяє системі зберігати рівномірний розподіл частинок в об’ємі, тобто запобігати дії сил тяжіння і процесам осідання або випливання частинок. Основними умовами цієї стійкості є висока дисперсність і участь частинок дисперсної фази у броунівському русі. Агрегативна стійкість дисперсних систем – це здатність протистояти агрегації частинок. У цьому відношенні дисперсні системи поділяють на два класи: 1) термодинамічно стійкі, або ліофільні колоїди, які самочинно диспергуються та існують без додаткової стабілізації (мінеральні розчини поверхнево-активних речовин, розчини високомолекулярних речовин і т. п.). При утворенні цих систем вільна енергія Гіббса зменшується (∆G<0); 2) термодинамічно нестійкі або ліофобні системи (золі, суспензії, емульсії). Для них ∆G>0. Уявлення про седиментаційну і агрегативну стійкість у даний час доповнює поняття про конденсаційну (фазову) стійкість. Мається на увазі структура і міцність агрегатів, які утворюються при коагуляції дисперсної системи. Конденсаційно стійкі системи утворюють нещільні агрегати (флокули) або пухкі осади. У них часточки втрачають свою індивідуальну рухливість, але зберігаються як такі протягом тривалого часу. Цьому сприяють прошарки дисперсного середовища між частинками дисперсної фази. Агрегати з такою структурою при відповідних умовах можуть знову розпадатися на окремі частинки, тобто зазнають пептизації. Конденсаційно нестійкі системи характеризуються утворенням агрегатів з міцною структурою. Для цього проводять безпосередні фазові контакти частинок одна з одною, процеси кристалізації, зрощування частинок і т. п. Утворення таких структур є необоротним. Фактори стійкості дисперсних систем. Агрегативна стійкість дисперсних систем досить різноманітна. Одні системи можуть існувати секунди після їх утворення, інші досить довговічні. Найбільш нестійкими за своєю природою є гідрофобні колоїдні системи, для яких характерна слабка взаємодія між частинками дисперсної фази і дисперсного середовища. Для надання стабільності таким системам необхідна присутність певних факторів стійкості. Фактори агрегативної стійкості дисперсних систем поділяються на термодинамічні та кінетичні. До термодинамічних факторів належать наступні: 1) електростатичні – сприяють створенню електростатичних сил відштовхування, які зростають при збільшенні потенціалу поверхні частинок (φ) і особливо електрокінетичного (ξ) потенціалу; 2) адсорбційно-сольватний – зумовлює зменшення міжфазового натягу і зниження енергії Гіббса поверхні поділу; 3) ентропійний – є додатковим до двох перших факторів і діє у високодисперсних системах, частинки дисперсної фази яких беруть участь у броунівському русі; сприяють рівномірному розподілу частинок по всьому об’єму системи. До кінетичних факторів стійкості належать: 1) структурно-механічний – зв’язаний з утворенням на поверхні частинок захисних шарів (плівок), які володіють пружністю і механічною міцністю та стійкістю до руйнування; 2) гідродинамічний – понижує швидкість агрегації внаслідок зміни в’язкості середовища, густини дисперсної фази і дисперсного середовища. У реальних системах агрегативна стійкість обумовлюється одночасною дією декількох факторів. При цьому основну роль відіграють два фактори агрегативної стійкості: електростатичний бар’єр, що створюється силами відштовхування, і адсорбційно-сольватний бар’єр. Теорії стійкості і коагуляції. Всі теорії коагуляції гідрофобних колоїдів в основному можна розділити на адсорбційні та електростатичні. Адсорбційну теорію коагуляції описав Г.Фрейндліх. Вона базується на тому, що при коагуляції золів йони-коагулятори адсорбуються колоїдними частинками відповідно до ізотерми адсорбції. При цьому коагуляція настає при однаковому зниженні ξ-потенціалу, але досягається при адсорбції еквівалентних кількостей різних йонів. Ця теорія пояснила зниження ξ-потенціалу до критичного значення зменшенням числа зарядів потенціалоутворюючих йонів внаслідок нейтралізації їх адсорбованими йонами-коагуляторами. Але ця теорія виявилася обмеженою у використанні, бо далеко не завжди спостерігається еквівалентність адсорбції різних електролітів і співвідношення ізотерм адсорбції різних йонів. Крім того, у багатьох випадках коагуляція пов’язана зі зміною лише у дифузному шарі, а заряд потенціалоутворюючих йонів залишається сталим. Електростатичну теорію розробив Г.Мюллер. На відміну від адсорбційної, ця теорія базувалася на тому, що введення електроліту в золь не змінює загального заряду у подвійному шарі частинки, а викликає стискання дифузного шару. Зменшення товщини йонної атмосфери призводить до зниження ξ-потенціалу, яке можна вирахувати на основі теорії сильних електролітів Дебая-Хюккеля. Внаслідок зниження ξ-потенціалу зменшується стабільність золю. Сучасну теорію стійкості колоїдних систем розробили Б.Дерягін, Л.Ландау (1937), Е.Фервей, Я.Овербек (1941). За першими літерами прізвищ авторів теорія отримала назву ДЛФО. Згідно з цією теорією, між частинками при їх зближенні виникає розклинювальний тиск, який розділяє рідкий прошарок в результаті дії сил притягання і відштовхування. Стан системи залежить від балансу енергії притягання і енергії відштовхування. Перевага енергії відштовхування призводить до стійкості системи. Перевага енергії притягання викликає порушення агрегативної стійкості, тобто коагуляцію. Зміну енергії взаємодії між двома частинками при їх зближенні можна зобразити графічно (рис. 7.12). На осі абсцис відкладають віддаль між частинками. Енергію відштовхування приймають за позитивну і відкладають на осі ординат вверх від початку координат. Зміну цієї енергії з віддалю виражає крива 1. Енергію притягання, як від’ємну, відкладають вниз від осі абсцис. Її залежність від віддалі виражає крива 2. Сумарну енергію системи із двох частинок одержують сумуванням енергії відштовхування і енергії притягання:
де: Uвідшт – енергія відштовхування; Uприт – енергія притягання; В – множник, який залежить від значень електричних потенціалів ДЕС, властивостей середовища, температури; е – основа натурального логарифма; χ – величина, зворотна товщині дифузного шару; h – віддаль між частинками; А – константа молекулярних сил притягання.
Рис. 7.12. Потенційні криві взаємодії колоїдних частинок: 1 – енергія відш-товхування; 2 – енергія притягання; 3 – результуюча крива. Природа сил притягання і сил відштовхування різна, тому залежності енергії притягання і енергії відштовхування від віддалі мають різний характер. Енергія притягання обумовлена вандерваальсовими силами і змінюється обернено пропорційно квадрату віддалі між частинками. Сили відштовхування, згідно з теорією ДЛФО, мають електростатичний характер. Вони проявляються тоді, коли дві однойменно заряджені частинки зближуються настільки, що їх дифузні шари взаємно перекриваються (рис. 7.12, а). Енергія відштовхування зменшується з віддалю за експоненційним законом. Оскільки доданок розклинювального тиску має різні знаки (Uвідшт>0, Uпритяг<0), то знак сумарного розклинювального тиску залежить від якоїсь із взаємодій. Для визначення стану даної системи проводять розрахунок сумарної енергії на різних віддалях h шляхом алгебраїчного складання ординат кривих 1 і 2. За даними розрахунків будують результуючу криву (рис 7.12). Аналіз результуючої кривої дозволяє виділити на ній характерні ділянки: в зоні малих віддалей на кривій є глибокий первинний мінімум (потенціальна яма), що вказує на значне переважання енергії притягання. В зоні великих віддалей також є деяка перевага енергії притягання, що відображається вторинним неглибоким мінімумом (друга потенціальна яма). В зоні середніх віддалей на кривій є максимум, і якщо він розташований над віссю абсцис, то проявляється потенціальний енергетичний бар’єр сил відштовхування ∆Uб. Величина ∆Uб зв’язана з агрегативною стійкістю системи. Частинки дисперсної фази мають певну кінетичну енергію (kT), за рахунок якої вони можуть зближуватися на ту чи іншу віддаль. Залежно від висоти бар’єра і глибини потенціальних ям можливі наступні варіанти поведінки частинок при зближенні: 1) високий енергетичний бар’єр (∆Uб>>kТ) і відсутність або неглибокий вторинний мінімум (∆Uя<<kТ) означають, що частинки не можуть подолати бар’єр і розходяться без взаємодії. У цьому випадку система агрегативно стійка (рис. 7.13, б); 2) при малій висоті бар’єра і незначному вторинному мінімумі, коли ∆Uб ≈ ∆Uя<<kТ, броунівський рух може зблизити частинки до таких мінімальних віддалей, що вони попадуть у першу потенціальну яму, при цьому частинки вступають у ближню взаємодію, тобто безпосередньо стикаються і проходить коагуляція (рис. 7.13, в); 3) при помірно глибокому вторинному мінімумі (∆Uя>>kТ) і наявності помітного енергетичного бар’єра
Рис. 7.13. Схема взаємодії колоїдних частинок: а – перикриття дифузних шарів; б – агрегативно стійка система; в – коагуляція. Наведені закономірності добре узгоджуються з поведінкою гідрофобних золів. Якщо частинки золю мають високий електричний потенціал і достатню товщину дифузного шару, то при перекриванні подвійного електричного шару двох частинок енергія електростатичного відштовхування переважає над енергією міжмолекулярного притягання. Виникає енергетичний бар’єр, він перешкоджає злипанню частинок. Частинки віддаляються одна від одної і система є агрегативно стійка (рис. 7.13, б). Стискання дифузного шару, наприклад, при додаванні електролітів, призводить до того, що віддаль h (рис. 7.13, а) між твердими частинками виявляється дуже малою. На цій віддалі енергія притягання значно перевищує енергію відштовхування. За таких умов енергетичний бар’єр незначний і система агрегативно нестійка, тому золь коагулює (рис. 7.13, в). Таким чином, із розглянутих можливих випадків взаємодії частинок випливає, що дисперсна система агрегативно стійка тільки при високому енергетичному бар’єрі сил відштовхування. Тому всі ті фактори, які знижують величину енергетичного бар’єра ∆Uб неминуче послаблюють агрегативну стійкість системи.
§9. Коагуляція гідрофобних золів. Золі, суспензії, емульсії, як ліофобні дисперсні системи, є агрегативно нестійкими внаслідок надлишку поверхневої енергії Гіббса. Процес укрупнення частинок (коагуляція) відбувається самовільно, бо супроводжується зменшенням питомої поверхні і зниженням енергії Гіббса. При коагуляції змінюються фізико-хімічні властивості систем: з’являється каламутність, знижується осмотичний тиск, змінюються електропровідність і характер в’язкості. Фактором, що викликає коагуляцію, може бути будь-який агент, який порушує агрегативну стійкість системи, наприклад температурні зміни (сильне нагрівання або охолодження аж до заморожування), механічні втручання (перемішування, перекачування, інтенсивне струшування), дія світла й інших випромінювань, електричні розряди. Однак, найвагомішим фактором є дія електролітів. Введення їх в золі дуже швидко призводить до зміни товщини подвійного електричного шару і ξ-потенціалу, який є одним з головних факторів стійкості гідрофобних колоїдних систем. Коагуляція під дією електролітів. Правило Шульце-Гарді. Коагуляція відбувається при певній кількості доданого електроліту. Мінімальну молярну концентрацію еквівалента електроліту (в моль/л або ммоль/л), яка викликає за певний проміжок часу коагуляцію колоїдного розчину, називають порогом коагуляції – ск. Значення ск є порівняльним критерієм агрегативної стійкості дисперсних систем. Величина ск визначається зарядом йона – коагулятора. Знак заряду йона-коагулятора протилежний знаку заряду гранули. Величину, обернену порогу коагуляції, називають коагулюючою здатністю і позначають Vк Спостереження Г.Шульце (1882) показали, що коагулюючою здатністю володіє один із йонів електроліту (йон-коагулятор). Причому, коагулююча здатність йона-коагулятора зростає зі збільшенням його заряду (правило Шульце). Дещо пізніше М.Гарді (1900) довів, що заряд коагулюючого йона завжди протилежний заряду колоїдної частинки (правило Гарді). Таким чином, коагуляцію від’ємно зарядженого золю викликають катіони доданого електроліту. Для золю з позитивно зарядженими частинками йонами-коагуляторами є аніони. Закономірність, відкрита Шульце і Гарді, як правило Шульце-Гарді звучить так: коагулюючою дією володіє той йон електроліту, який має заряд, протилежний заряду гранули; коагулююча дія тим сильніша, чим вищий заряд йона-коагулятора (правило значності). Згідно з правилом Шульце-Гарді, поріг коагуляції йона-коагулятора ск зменшується зі збільшенням його заряду. Для двозаряджених йонів поріг коагуляції в десятки, а тризарядних – у сотні разів менший, ніж для однозарядних: с¢к:с¢¢к:с¢¢¢к = 500:25:1. Підвищення коагулюючої дії йона зі зростанням його заряду пояснюється сильнішим ефектом стиснення подвійного електричного шару йонами з вищим зарядом. У ряді неорганічних йонів з однаковим зарядом їх коагулююча активність збільшується зі зменшенням радіуса гідратованого йона. Так, у ряді неорганічних катіонів і аніонів коагулююча активність та ступінь гідратації змінюється в ліотропних рядах, або рядах Гофмейстера: ступінь гідратації ступінь гідратації В гомололічних рядах електролітів з органічними йонами коагулююча здатність, згідно з правилом Траубе, рівномірно збільшується пропорційно до збільшення числа метиленових груп (-СН2-). Отже, в ряді органічних йонів коагулююча дія зростає зі збільшенням адсорбційної здатності. Розрізняють два види коагуляції колоїдних розчинів електролітами: концентраційну і нейтралізаційну. Концентраційна коагуляція пов’язана зі збільшенням концентрації електроліту, який не вступає в хімічну взаємодію з компонентами колоїдного розчину, а також не спроможний до специфічної адсорбції. Такі електроліти називаються індеферентними. Додавання таких електролітів зумовлює електростатичний ефект стиснення подвійного електричного шару, а отже і зменшення ξ-потенціалу. Стиснення дифузного шару є наслідком двох причин: а) переміщення частини протийонів дифузного шару в адсорбційний, що веде до додаткового зниження ξ-потенціалу; б) пригнічення дифузії протийонів і зменшення розпушеності дифузного шару. Цей фактор є переважаючим для систем з сильнозарядженими частинками. Нейтралізаційна (адсорбційна) коагуляція є наслідком зменшення потенціалу твердої поверхні і відбувається у колоїдних системах зі слабозарядженими частинками. Цей вид коагуляції викликають електроліти з йонами, які здатні до специфічної адсорбції на поверхні частинок і заряджені протилежно до них. Процес коагуляції ускладнюється, якщо використовують суміш електролітів. Відбувається зміщення адсорбційної рівноваги, яка супроводжується перерозподілом йонів подвійного шару і зміною порогів коагуляції. При цьому спостерігаються такі явища: адитивність, антагонізм та синергізм дії електролітів. Адитивність (від лат. additivus – придатковий) проявляється при сумуванні коагулюючої дії електролітів. Якщо побудувати графік, на осі абсцис якого відкласти концентрацію одного електроліту, прийняту за 100% і необхідну для швидкої коагуляції золю у відсутності другого електроліту, а на осі ординат те ж саме для другого електроліту, то адитивна дія електролітів відобразиться прямою (рис. 7.14.1). Це означає, що для початку коагуляції можна взяти 100% порогової концентрації (ск) одного чи другого електроліту або суміші із 50% порогових концентрацій одного і другого електролітів.
Рис. 7.14. Коагуляція су-міші електролітів: 1 – адитивність коагуляцій-ної дії, 2 – антагонізм, 3 – синергізм. Антагонізмом електролітів називають явище, при якому для початку швидкої коагуляції вимагається суміші електролітів більше, ніж для кожного з них зокрема. Такій залежності відповідає крива 2 (рис. 7.14). Синергізм (від грецьк. діючий разом) електролітів проявляється у ефективнішій дії суміші, ніж кожного електроліту зокрема. Для цього випадку характерна крива 3 (рис. 7.14). Гетерокоагуляція. Взаємна коагуляція колоїдів. Взаємна коагуляція спостерігається при змішуванні золів із різнойменно зарядженими частинками. Механізм взаємодії коагуляції в тому, що при перекриванні подвійних шарів колоїдних частинок з різними знаками відбувається не відштовхування, а електростатичне притягання і швидка агрегація частинок. Явище взаємної коагуляції має важливе значення у процесах ґрунтоутворення та очищенні води. Явище звикання золів. У цілому ряді випадків коагуляція залежить від способу додавання електроліту-коагулятора. Якщо електроліт додавати невеликими порціями, то коагуляція настає при вищій сумарній його концентрації, ніж при одноразовому внесенні великої кількості. Це явище називають звиканням золю. Причиною звикання золів може бути утворення пептизатора або адсорбція йонів, заряджених однойменно з частинкою, що призводить до збільшення заряду частинок. Біологічне значення коагуляції. Коагуляція широко розповсюджена у природі. Вона лежить в основі утворення атмосферних опадів, в ґрунтових процесах. Так, у дельтах рік відбувається коагуляція ґрунтових колоїдів морською водою, в результаті утворюються родючі ґрунти. Коагуляція використовується для очищення природної і стічної води, боротьби з забрудненням повітряного простору аерозолями. Коагуляція лежить в основі багатьох технологічних процесів, які пов’язані з одержанням і очищенням харчових продуктів (цукрози, сирів, коров’ячого масла та ін.). Згортання крові, лімфи, звертання молока, травлення їжі і кормів базується на процесах коагуляції. За допомогою коагуляції із розчинів одержують білки, нуклеїнові кислоти та інші біополімери. Реакція коагуляції дозволяє визначити групи крові у людини і тварин.
КОНТРОЛЬНІ ЗАВДАННЯ 1. Що таке дисперсні системи? Їх класифікація. 2. Методи одержання колоїдних систем. 3. В чому суть диспергаційних методів одержання колоїдних систем? 4. На яких принципах ґрунтуються конденсаційні методи одержання колоїдних систем? 5. Що таке пептизація? 6. Що таке діаліз? Типи. 7. Поняття про ультрафільтрацію. 8. Що таке ліофобні і ліофільні колоїди? 9. Поняття про компенсаційний діаліз. Вівідіаліз. 10. Чим викликаний броунівський рух колоїдних систем? Чому броунівський рух відсутній у мікрогетерогенних системах? 11. Явище дифузії і осмотичний тиск колоїдних систем. Від яких факторів залежить величина осмотичного тиску у колоїдних системах? 12. Написати рівняння, за допомогою якого розраховується швидкість седиментації. 13. В чому суть явища Тіндаля? 14. Написати рівняння Релея і пояснити його зміст. 15. Принцип роботи ультрамікроскопа. 16. Що таке опалесценція? У яких системах вона спостерігається? 17. Принципи роботи електронного мікроскопа. 18. Будова міцели гідрофобного золю. Навести конкретний приклад. 19. Що таке потенціаловизначаючі йони і протийони? 20. Дати загальну характеристику електрокінетичним явищам. 21. Як виникають електрокінетичні потенціали (ξ-потенціали)? 22. Написати рівняння Гельмгольца-Смолуховського. 23. Що таке електроосмос? 24. Поняття про електрофорез. Його значення у біології. 25. Дати характеристику потенціалам перебігу і седиментації. 26. Види стійкості гідрофобних золів. 27. Дати характеристику седиментаційної стійкості колоїдних систем. 28. Агрегативна стійкість колоїдних систем. 29. Що таке конденсаційна стійкість? 30. Класифікація факторів стійкості дисперсних колоїдних систем. 31. Дати загальну характеристику електростатичним адсорбційно-сольватним і ентропічним факторам стійкості. 32. Теорія Г.Фрейндліха, Г.Мюллера та ДЛФО. 33. Коагуляція гідрофобних золів. Фактори, які зумовлюють коагуляцію. 34. Коагуляція під впливом електролітів. Правило Шульце-Гарді. 35. Що називають порогом коагуляції? 36. Якою формулою виражається товщина подвійного електричного шару? 37. Чим обумовлена різниця порогів коагуляції електролітів, які містять йони-коагулятори однакової валентності? 38. Що проходить при перекритті йонних атмосфер колоїдних частинок? 39. Які види коагуляції відбуваються під впливом електролітів? 40. Дати характеристику концентраційної коагуляції. 41. Що таке нейтралізаційна (адсорбційна) коагуляція? 42. Поняття про взаємну коагуляцію колоїдних систем. 43. Механізм коагуляції сумішшю електролітів. 44. Біологічне значення коагуляції.
Розділ VIII До високомолекулярних сполук (ВМС) належать сполуки з молекулярною масою порядку 104-106 і вище. Вони утворюються в результаті поліконденсації або полімеризацій невеликих молекул (мономерів) – моноцукориди, ненасичені вуглеводні, амінокислоти, ненасичені кислоти і т. п. В результаті цих процесів утворюються довгі ланцюги розгалуженої, нерозгалуженої, циклічної форм. Так, наприклад, до ВМС наледать поліпептиди:
Розчини ВМС володіють рядом властивостей, подібних до колоїдних розчинів: повільно дифундують, не проникають через діалізаційні мембрани, розмір частинок відповідає колоїдним (1-100 нм). Однак, на відміну від колоїдів, у розчинах ВМС відсутня межа поділу; крім того, вони здатні самочинно розчинятися у відповідних рідинах, не вимагаючи для цього стабілізаторів чи затрати енергії ззовні. Розчини ВМС досить стійкі, а відсутність межі поділу, незважаючи на великий розмір частинок, пояснюється тим, що частинки ВМС є своєрідним у просторовому відношенні “клубком” дуже довгих ланцюгів. Товщина цих ланцюгів не перевищує товщини 1 молекули, що, незважаючи на велику довжину, виключає межу поділу і наближує ці розчини за властивостями до істинних. Високомолекулярні сполуки мають і специфічні властивості, вони набрякають, їх розчини володіють високою в’язкістю і здатністю до драгління. Легкість розчинення ВМС і стійкість їх розчинів обумовлені наявністю у їх структурі великої кількості ліофільних груп, тобто груп, що володіють високою спорідненістю за розчинниками. Ця властивість стала основою для поділу колоїдних розчинів на ліофільні і ліофобні. Більшість ВМС належать до ліофільних речовин. У їх структурі велика кількість ліофільних груп. Спонслер встановив, що одна група -ОН притягує три молекли води,
§1. Класифікація і структура високомолекулярних сполук. Молекули ВМС надзвичайно великі. Природні ВМС (біополімери) харктеризуються постійним значенням молекулярної маси. На відміну від них, синтетичні полімери є полідисперсними системами, тому що складаються з суміші макромолекул різної довжини і маси. Тому молекулярна маса таких полімерів є середнє значення М. Усі найважливіші властивості ВМС пов’язані з їх будовою. Розрізняють три основних типи структури ланцюгів: лінійну, розгалужену, просторову. Лінійні полімери (каучук) побудовані із довгих ланцюгів одномірних елементів (рис. 8.1, а). Розгалужені полімери мають ланцюги з боковими відгалуженнями (рис. 8.1, б). Так побудовані молекули крохмалю. Просторові полімери є трирозмірною сіткою (рис. 8.1, в), яка утворюється при сполученні відрізків ланцюгів хімічними зв’язками (наприклад, фенолформальдегідні смоли). Із просторових полімерів в особливу групу виділяють полімери зі зшитою структурою, ланцюги яких сполучені короткими містковими хімічними зв’язками за допомогою атомів Оксигену або Сульфуру (рис. 9.1, г). Таку структуру має, наприклад, гума. Специфічні властивості полімерів обумовлені, головним чином, двома особливостями: 1) існуванням двох типів зв’язків – хімічних і міжмолекулярних, які утримують макромолекулярні ланцюги один біля одного; 2) гнучкістю ланцюгів, пов’язаною з внутрішнім обертанням ланок.
Рис. 8.1. Схема будови макромолекул поліме-рів: а – лінійного; б – розгалуженого; в – прос-торового; г – зшитого.
§2. Властивості розчинів високомолекулярних сполук. ВМС можуть утворювати як істинні, так і колоїдні розчини (дисперсії). Характер одержуваного розчину ВМС залежить від спорідненості їх з розчинником. Якщо полярність розчинника відповідає полярності ВМС, то відбувається істинне розчинення з утворенням молекулярних розчинів (желатина у воді). Невідповідність полярності розчинника і ВМС веде до утворення золів або дисперсій. До основних властивостей розчинів ВМС належить: набрякання, стійкість, в’язкість, тиск набрякання, колоїдний захист, відмокання. Набрякання. Істинному розчиненню полімерів часто передує процес набрякання – самочинний процес проникнення молекул розчинника між молекули ВМС, що веде до значного збільшення об’єму і маси ВМС. При контакті полімеру з розчинником відбувається взаємна дифузія молекул розчинника в полімер, а макромолекул полімеру – в розчинник. Однак, швидкість дифузії в обидвох напрямках буде відрізнятись у тій же пропорції, що і розміри та рухливості дифундуючих молекул. Велика різниця в рухливості молекул розчинника і молекул ВМС є причиною набрякання. Мірою кількісної характеристики набрякання є ступінь набрякання α, який може виражатися в об’ємних і масових величинах:
де V0 і V, m0 і m – відповідно об’єми і маси вихідного і набряклого полімеру. Виходячи із структури полімеру і температури, набрякання може бути обмежене і необмежене. На рис. 8.2 наведені кінетичні криві для обох випадків. При обмеженому набряканні (крива 1) α досягає найбільшого значення, після набрякання не залежить від часу (набрякання желатини у холодній воді). Для необмеженого набрякання характерна залежність (крива 2), яка проходить через максимум і зменшується до нуля в результаті поступового розчинення полімеру (желатина в гарячій воді).
Рис. 8.2. Кінетичні криві набрякання: 1 – обмежене набрякання; 2-необмежене набрякання. Обмеженість чи необмеженість набрякання визначається співвідношенням енергій зв’язків у полімері з енергією сольватації і ентропійним фактором. В лінійних і розгалужених полімерах макромолекули сполучені вандерваальсовими силами, енергія яких невелика, тому енергія сольватації та ентропійний фактор вже при 20ºС перевищують їх. За таких умов набрякання проходить необмежено. Якщо між ланцюгами полімеру існує хімічний зв'язок, то для його розриву часто недостатньою є енергія сольватації і ентропійного фактора. Набрякання відбувається обмежено, і полімер драгліє. Набрякання не можна вважати чисто фізичним явищем, при якому молекули розчинника механічно проникають у простори між ланцюгами полімеру. В основі процесу набрякання лежить сольватація макромолекулярних ланцюгів. Про це свідчить виділення теплоти набрякання і контракція (зменшення загального об’єму системи). Причиною контракції є орієнтація молекул розчинника у сольватних шарах. Процес набрякання включає дві стадії. На першій відбувається виділення теплоти набрякання ΔН, спостерігається контракція системи, але α не досягає високих значень. Залежність між ΔН і α виражає емпірична формула:
де: а і в – константи. Друга стадія майже не супроводжується контракцією і виділенням теплоти, зате відбувається збільшення ступеня набрякання (α) і об’єму набряклого полімеру. Самовільне набрякання і розчинення полімеру супроводжується зменшенням енергії Гіббса (ΔG<0), а це можливо у двох випадках: 1. Якщо розчинення відбувається з виділенням теплоти і ΔН<0. Цей випадок має місце при розчиненні полярних полімерів у полярних розчинниках. Енергія сольватації полярних груп полімера більша, ніж затрати енергії на переборювання сил зчеплення макромолекул. 2. За умови, що ΔS>0. Для процесів розчинення зміни ентропії завжди позитивні. Велике значення величини ΔS призводить до розщеплення деяких полімерів навіть з поглинанням тепла, оскільки енергія може зменшуватися за рахунок ентропійного фактора. Тиск набрякання. Набрякання полімерів веде до збільшення об’єму у 10-15 разів і при цьому виникає тиск набрякання, який досягає сотень мегапаскалів. Тиск набрякання особливо великий при поглинанні полімером перших пропорцій розчинника, потім він зменшується, а при досягненні рівноваги між полімером і розчинником падає до нуля. Цей тиск можна визначити за рівнянням: рH = Kcn, (8.3) де: К та n – константи; с – концентрація сухої ВМС в 1 см2 системи, яка утворилася при набряканні. Це рівняння подібне до емпіричного рівняння адсорбції Фрейндліха. Фактори стійкості розчинів ВМС. Розчини ВМС у добре розчинюваних їх рідинах агрегативно стійкі. Порушити стійкість розчинів полімерів можна шляхом погіршення їх розчинності – введенням електролітів або рідин, які погано розчиняють цей полімер. Так, наприклад, для білків нерозчинниками є етанол, пропанон. У присутності електролітів і нерозчинників проходить процес виділення ВМС із розчину – висолювання. Цей процес подібний до коагуляції, однак, якщо для коагуляції золів вимагаються малі кількості електроліту і процес коагуляції необоротний, то для руйнування розчину ВМС необхідна висока концентрація електроліту. При цьому відбувається оборотний процес і спостерігається невиконання правила Шульце-Гарді. В основі механізму висолювання ВМС лежить процес дегідратації. Йони введеного електроліту чи молекули спирту ніби “забирають” велику частину розчинника від макромолекул ВМС. Концентрацію електроліту, при якій настає швидке осадження полімеру, називають порогом висолювання ВМС. Висолююча дія електролітів залежить від здатності йонів цих електролітів до гідратації. Гофмейстером була встановлена така послідовність висолюючої дії йонів (ліотропний ряд, або ряд Гофмейстера): SO42–>F–>C6H5O73–>C4H4O62–>C2H4O2–>Cl–>NO3–>Br–>CNS–. Лівіше Cl– розташовані йони, які знижують стійкість розчинів ВМС, а йони NO3–, Br–, J–, CNS–, навпаки, підвищують їх стійкість. Це пояснюється тим, що йони, розташовані зліва, добре гідратуються, віднімаючи масу води від колоїдних частинок чи розчинів ВМС, а ті, що справа, самі ж адсорбуються на них, збільшуючи їх заряд і водну оболонку. Катіони також утворюють ліотропний ряд: Li+>Na+>K+>Rb+>Cs+>Mg2+>Ca2+>Sr2+>Ba2+. Висолювання білків концентрованими розчинами солей є одним з головних методів фракціонування білкових сумішей на альбуміни і глобуліни. Зниження розчинності білків можна досягти додаванням спирту, солей та охолодженням. На цьому базуються методи детального фракціонування білкових сумішей, наприклад, з сироватки крові за допомогою цього методу вилучено понад двадцять білків. Специфічне необоротне осадження білків називають денатурацією. Вона відбувається під впливом зміни температури, при дії сильних кислот та лугів, ультразвуку, високого тиску, променевої енергії. При денатурації порушується форма і розміри молекул, змінюється оптична активність білків, збільшується в’язкість розчинів, тому що глобулярна форма розкручується з утворенням ниткоподібних молекул, зменшується розчинність білків і ступінь набрякання, відбувається зняття з колоїдних частинок електричного заряду та ін. За Жолі, денатурація – це будь-яка модифікація вторинної, третинної або четвертинної структури білкової молекули, за винятком розриву ковалентних зв’язків. Денатурація викликає зміни специфічних біологічних властивостей білків – втрату ферментативної та імунологічної активності. Колоїдний захист. Суміш ВМС і колоїдів часто проявляє особливі властивості. У випадку переваги у суміші полімеру (білка), він адсорбується на поверхні колоїдної частинки, утворюючи крупний агрегат з гідрофільними властивостями. Стійкість його буде середньою між обидвома видами взаємодіючих частинок. Це явище називається захистом золю високомолекулярними сполуками (колоїдний захист). Здатність ВМС захищати золь золота від коагуляції електролітом вимірюють золотим числом, тобто кількістю міліграмів сухого полімеру (наприклад, желатини), яка захищає 10 мл червоного гідрозолю золота від коагуляції 1 мл 10%-го розчину NaCl. Для золю гідроксиду заліза існує залізне число, для золю срібла – срібне число і т. п. (табл. 8.1). Таблиця 8.1. Характеристика захисної здатності деяких ВМС. ВМС Число золоте срібне залізне Желатина 0,01 0,035
|
||
|
|
Последнее изменение этой страницы: 2024-06-27; просмотров: 73; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.134 (0.056 с.) |

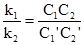 .
.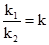 і
і 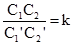 . (5.13)
. (5.13)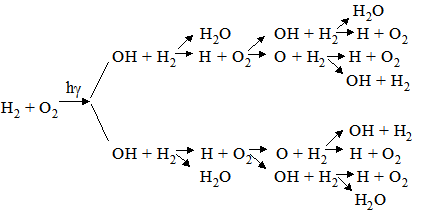 .
.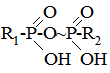
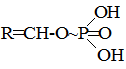
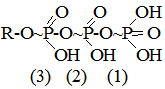
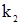
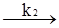 D+K. (2)
D+K. (2)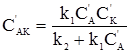 .
.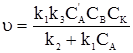 .
.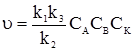 ;
;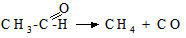 , яку каталізує пароподібний Йод. У відсутності пари Йоду енергія активації становить 191,0 кДж/моль, в її присутності – 136,0 кДж/моль. Константа швидкості зростає у 10000 разів. Це відбувається тому, що реакція відбувається у дві стадії:
, яку каталізує пароподібний Йод. У відсутності пари Йоду енергія активації становить 191,0 кДж/моль, в її присутності – 136,0 кДж/моль. Константа швидкості зростає у 10000 разів. Це відбувається тому, що реакція відбувається у дві стадії: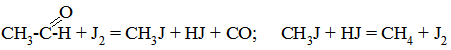 .
.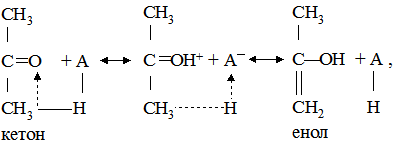
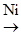 СН4+2Н2О.
СН4+2Н2О.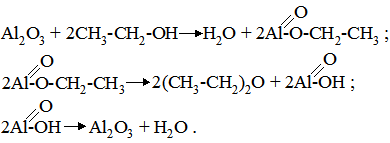
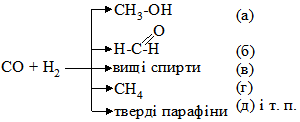
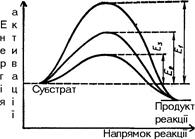
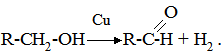
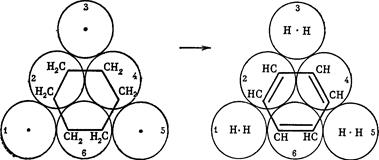
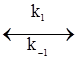 FS
FS 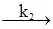 P+F,
P+F,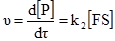 . (5.15)
. (5.15)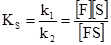 . (5.18)
. (5.18)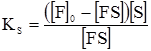 , (5.19)
, (5.19)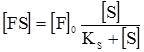 . (5.20)
. (5.20)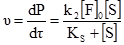 . (5.21)
. (5.21)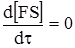 .
.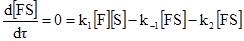 , а згідно з (5.16):
, а згідно з (5.16):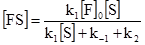 . (5.23)
. (5.23)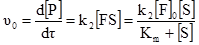 , (5.24)
, (5.24)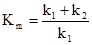 . (5.25)
. (5.25)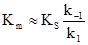 .
.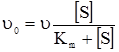 . (5.27)
. (5.27)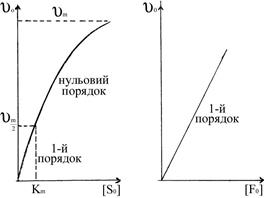
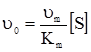 . При високій концентрації субстрату [S]>>Кm швидкість має нульовий порядок відносно [S]: υ0 ≈ υm. Ці два випадки стосуються як початкової фази, так і наступних періодів реакції (рис. 5.3, а).
. При високій концентрації субстрату [S]>>Кm швидкість має нульовий порядок відносно [S]: υ0 ≈ υm. Ці два випадки стосуються як початкової фази, так і наступних періодів реакції (рис. 5.3, а).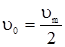 рівняння (5.27) дає:
рівняння (5.27) дає: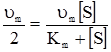 , або Km=[S].
, або Km=[S].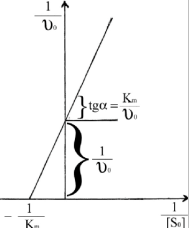
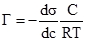 , (6.1)
, (6.1) – зміна поверхневого натягу зі зміною концентрації поверхнево-активної речовини. Щоб виключити вплив концентрації на значення наведеної похідної, беруть її граничне значення (при с ® 0), тоді
– зміна поверхневого натягу зі зміною концентрації поверхнево-активної речовини. Щоб виключити вплив концентрації на значення наведеної похідної, беруть її граничне значення (при с ® 0), тоді 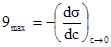 – поверхнева активність, Дж·м/моль або н·м2/моль.
– поверхнева активність, Дж·м/моль або н·м2/моль.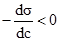 , а адсорбція (Г>0) буде мати додатне значення. Таку адсорбцію називають позитивною. Межею її є повне насичення поверхневого шару адсорбованою речовиною. Речовини з позитивною адсорбцією (жири, жирні кислоти, холестерол, кетони, більшість спиртів) називають також поверхнево-активними речовинами. Якщо ж
, а адсорбція (Г>0) буде мати додатне значення. Таку адсорбцію називають позитивною. Межею її є повне насичення поверхневого шару адсорбованою речовиною. Речовини з позитивною адсорбцією (жири, жирні кислоти, холестерол, кетони, більшість спиртів) називають також поверхнево-активними речовинами. Якщо ж 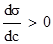 , то поверхневий натяг збільшується із зростанням концентрації розчиненої речовини, адсорбція (Г<0) буде мати від’ємне значення. Таку адсорбцію називають негативною.
, то поверхневий натяг збільшується із зростанням концентрації розчиненої речовини, адсорбція (Г<0) буде мати від’ємне значення. Таку адсорбцію називають негативною.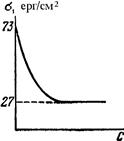
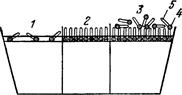
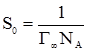 . (6.2)
. (6.2)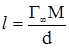 . (6.5)
. (6.5)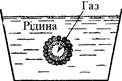
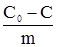
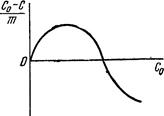
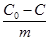

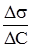 можна знайти як
можна знайти як 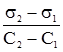 або як тангенс кута, утвореного дотичною до точки А на ізотермі поверхневого натягу з віссю абсцис. Підставивши знайдену величину поверхневої активності у рівняння Гіббса, можна вирахувати величину Г.
або як тангенс кута, утвореного дотичною до точки А на ізотермі поверхневого натягу з віссю абсцис. Підставивши знайдену величину поверхневої активності у рівняння Гіббса, можна вирахувати величину Г.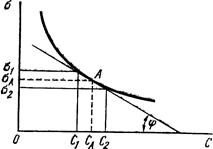
 AM.
AM.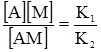 , (6.6)
, (6.6)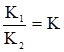 (константа адсорбційної рівноваги); тоді
(константа адсорбційної рівноваги); тоді 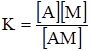 ; [AM] – кількість адсорбованих молекул газів; [A] – кількість вільних активних центрів, що відповідає числу молекул, які б могли бути адсорбовані; [M] – кількість неадсорбованих молекул.
; [AM] – кількість адсорбованих молекул газів; [A] – кількість вільних активних центрів, що відповідає числу молекул, які б могли бути адсорбовані; [M] – кількість неадсорбованих молекул.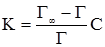 ;
;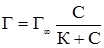 . (6.7)
. (6.7)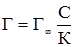 ,
,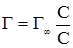 ,
,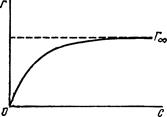
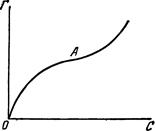
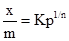 , або
, або 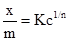 , (6.8)
, (6.8)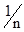 – емпіричний показник ступеня, необхідний для переведення рівняння в лінійну форму, що відображає ступінь кривизни ізотерми; K – питома адсорбція, якщо р = 1, то
– емпіричний показник ступеня, необхідний для переведення рівняння в лінійну форму, що відображає ступінь кривизни ізотерми; K – питома адсорбція, якщо р = 1, то 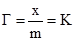 .
. 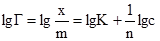 . (6.9)
. (6.9)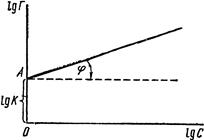
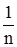 . Ізотерма адсорбції Фрейндліха виявилася неточною для малих і великих концентрацій адсорбтиву. Рівняння адсорбції Фрейндліха і відповідна йому парабола не описує прямолінійного зростання адсорбції при збільшенні концентрації адсорбтиву (у випадку низьких його концентраціях) і граничного значення адсорбції, коли подальше збільшення концентрації адсорбтиву не впливає на цей процес. Універсальнішою у цьому відношенні є ізотерма Ленгмюра і особливо БЕТ. Однак, для практичних цілей часто використовують рівняння ізотерми адсорбції Фрейндліха.
. Ізотерма адсорбції Фрейндліха виявилася неточною для малих і великих концентрацій адсорбтиву. Рівняння адсорбції Фрейндліха і відповідна йому парабола не описує прямолінійного зростання адсорбції при збільшенні концентрації адсорбтиву (у випадку низьких його концентраціях) і граничного значення адсорбції, коли подальше збільшення концентрації адсорбтиву не впливає на цей процес. Універсальнішою у цьому відношенні є ізотерма Ленгмюра і особливо БЕТ. Однак, для практичних цілей часто використовують рівняння ізотерми адсорбції Фрейндліха.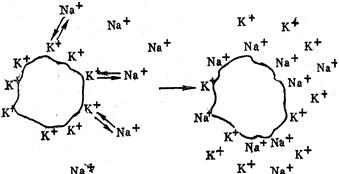
 ;
; 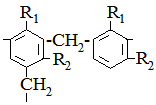 ,
,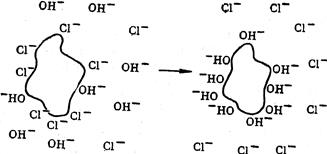
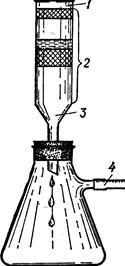


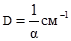 . (7.1)
. (7.1)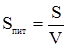 , (7.2)
, (7.2)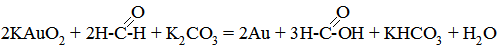 .
.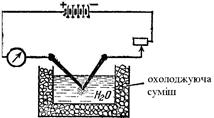
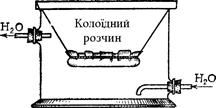




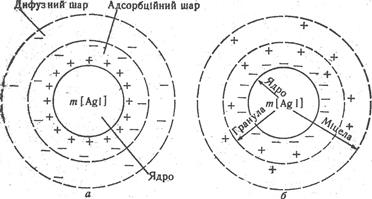
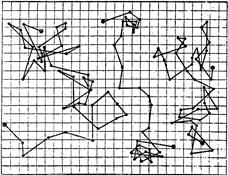
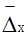 за час τ від коефіцієнта дифузії D виражається рівнянням Айнштайна:
за час τ від коефіцієнта дифузії D виражається рівнянням Айнштайна: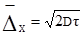 . (7.3)
. (7.3)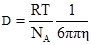 . (7.4)
. (7.4)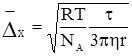 , (7.5)
, (7.5)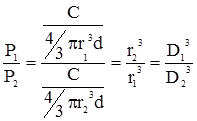 . (7.7)
. (7.7)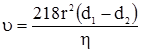 , (7.8)
, (7.8)

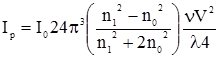 , (7.9)
, (7.9)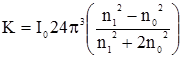 ,
,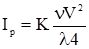 , (7.10)
, (7.10)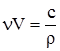
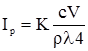 . (7.12)
. (7.12)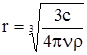 (7.14)
(7.14)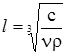 . (7.15)
. (7.15)
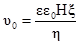 ,
,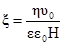 , (7.16)
, (7.16)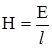 , Е – зовнішня різниця потенціалів, l – віддаль між електродами); ε – діелектрична стала рідини; ε0 – електрична стала, яка дорівнює 8,854·10–12
, Е – зовнішня різниця потенціалів, l – віддаль між електродами); ε – діелектрична стала рідини; ε0 – електрична стала, яка дорівнює 8,854·10–12  .
.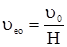 .
.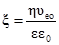 . (7.17)
. (7.17)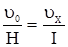 , (7.18)
, (7.18) . (7.19)
. (7.19)
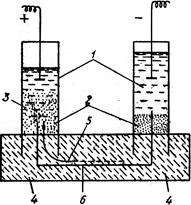
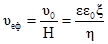 , (7.20)
, (7.20)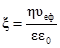 , якщо
, якщо 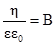 , де В = const, то
, де В = const, то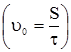 , а напруга зовнішнього електричного поля (градієнт потенціалу) – відношення зовнішньої різниці потенціалів Е до відстані між електродами l
, а напруга зовнішнього електричного поля (градієнт потенціалу) – відношення зовнішньої різниці потенціалів Е до відстані між електродами l 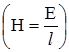 . На основі цього можна визначити електрофоретичну рухливість υеф:
. На основі цього можна визначити електрофоретичну рухливість υеф: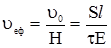 . (7.22)
. (7.22)
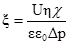 , (7.23)
, (7.23)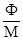 – електрична стала. Потенціал перебігу U визначають як різницю Uі–U0, де U0 – різниця потенціалів між електродами без тиску; Uі – різниця потенціалів при різних значеннях тиску. Визначення ξ-потенціалу цим методом дає вірогідніші дані, оскільки в експерименті немає необхідності накладати зовнішню різницю потенціалів, яка викликає побічні явища (поляризацію, нагрівання). При осіданні частинок дисперсної фази по висоті посудини виникає різниця потенціалів – потенціал седиментації. Причиною цього явища, оберненого електрофорезу, є також подвійний електричний шар, який деформується при терті осідаючих частинок об середовище. За величиною потенціалу седиментації можна розраховувати ξ-потенціал.
– електрична стала. Потенціал перебігу U визначають як різницю Uі–U0, де U0 – різниця потенціалів між електродами без тиску; Uі – різниця потенціалів при різних значеннях тиску. Визначення ξ-потенціалу цим методом дає вірогідніші дані, оскільки в експерименті немає необхідності накладати зовнішню різницю потенціалів, яка викликає побічні явища (поляризацію, нагрівання). При осіданні частинок дисперсної фази по висоті посудини виникає різниця потенціалів – потенціал седиментації. Причиною цього явища, оберненого електрофорезу, є також подвійний електричний шар, який деформується при терті осідаючих частинок об середовище. За величиною потенціалу седиментації можна розраховувати ξ-потенціал.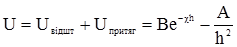 , (7.24)
, (7.24)
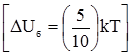 має місце взаємодія двох частинок, при якій вони не можуть розійтися (їх утримують сили притягання) і не можуть упритул наблизитися, бо цьому першкоджають сили відштовхування. За таких умов утворюються структуровані системи – драглі, у яких, однак, зберігаються прошарки середовища між частинками. Утворені драглі є періодичні колоїдні структури квазікристалічної будови.
має місце взаємодія двох частинок, при якій вони не можуть розійтися (їх утримують сили притягання) і не можуть упритул наблизитися, бо цьому першкоджають сили відштовхування. За таких умов утворюються структуровані системи – драглі, у яких, однак, зберігаються прошарки середовища між частинками. Утворені драглі є періодичні колоїдні структури квазікристалічної будови.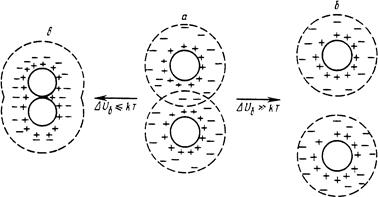
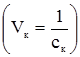 . Коагулюючу здатність Vк йона-коагулятора показує об’єм золю, скоагульованого 1 молем (або ммолем) йона-коагулятора.
. Коагулюючу здатність Vк йона-коагулятора показує об’єм золю, скоагульованого 1 молем (або ммолем) йона-коагулятора.


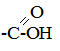 – чотири,
– чотири, 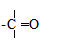 – дві, -NH2 – три молекули. Гідрофільність таких природних ВМС, як білки, поліцукориди, фосфатиди, обумовлена головним чином пептидними, етерними, подвійними, карбоксильними, спиртовими й амінними групами.
– дві, -NH2 – три молекули. Гідрофільність таких природних ВМС, як білки, поліцукориди, фосфатиди, обумовлена головним чином пептидними, етерними, подвійними, карбоксильними, спиртовими й амінними групами.
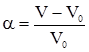 або
або 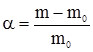 , (8.1)
, (8.1)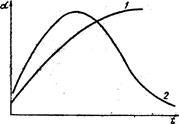
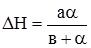 ,
,


