
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Будуючи графік залежності lgk від для тої чи іншої реакції, обчислюють тангенс кута нахилу прямої – і визначають величину енергії активації.Содержание книги
Поиск на нашем сайте
деяких електролітів. Електроліт Дифузний потенціал, В 0,1н KCl/0,001н KCl 0,1н NaCl/0,001н NaCl 0,1н KOH/0,001н KOH 0,1н NaOH/0,001н NaOH 0,1н HCl/0,001н HCl –0,0004 –0,0187 –0,0265 –0,0346 +0,0378
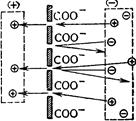
Дифузний потенціал може сильно зрости, якщо розчини електролітів різних концентрацій розділити мембраною, яка володіє проникністю тільки для йона з яким-небудь одним зарядом. Такі властивості деяких мембран пояснюють, зокрема, тим, що вільні карбоксильні групи цих мембран, заряджені негативно, притягують і пропускають тільки катіони і відштовхують аніони (рис. 4.11). Але існують також мембрани, проникні тільки для аніонів, наприклад оболонки еритроцитів. Можливо, що там вибірковість мембран якоюсь мірою обумовлена позитивно зарядженими аміногрупами.
Рис. 4.12. Мембранний по-тенціал, обумовлений не-гативним зарядом мембран Через мембрану (рис. 4.12) рухаються катіони, тому менш концентрований розчин заряджається позитивно. Величина потенціалу залежить від різниці концентрацій електроліту по обидва боки мембрани. Так, по обидва боки мембрани із колодію, що ділить 0,01 і 0,1М розчин КCl, виникає потенціал 45-55 мВ. У тканинах організму, навіть всередині однієї клітини, є мембранні потенціали, обумовлені морфологічною і хімічною неоднорідністю вмісту клітин. При роботі серця, скороченні м’язів виникають так звані струми дії. Існує теорія, яка пояснює їх виникнення як результат неоднакової проникності клітинних мембран для різних йонів. Внаслідок цього концентрація йонів по обидва боки мембран неоднакова. У момент збудження (скорочення м’язів) вибірковість проникності втрачається і крізь них направляється потік йонів – виникає електричний струм. Виникнення біоелектричних потенціалів пов’язане з тим, що у стані спокою електроліти всередині клітини вибірково зв’язуються білками. Виникає міжфазова різниця потенціалів між протоплазмою і водним розчином електроліту (потенціал спокою). При збудженні або пошкодженні клітини фазові властивості білків протоплазми змінюються, розподіл йонів стає іншим і відповідно змінюється потенціал (виникає потенціал дії або пошкодження). Існують і інші теорії виникнення біопотенціалів та біострумів, згідно з якими потенціали обумовлені нерівномірним розподілом йонів калію і хлору між зовнішнім та внутрішнім середовищем клітини (тобто є мембранними потенціалами) або між різними ділянками в одній клітині (наприклад, між поверхнями і прилягаючими до ядра шарами протоплазми). Цей потенціал належить до міжфазових і виникає як наслідок неоднакової адсорбційної здатності протоплазми клітин різних йонів.
§10. Полярографічний метод аналізу.
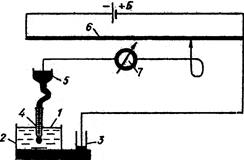
Рис. 4.13. Схема пристрою для по-лярографічного аналізу: 1 – аналі-зований розчин; 2 – шар ртуті; 3 – резервуар Меркурію; 4 – анод; 5 – краплинний електрод; 6 – гальва-нометр; 7 – рехорд. Метод, запропонований чеським вченим Я.Гейровським, базується на одержанні й аналізі кривих залежності струму в розчині від напруги, що поступово змінюється. Схему полярографа, за допомогою якого одержують криві, подано на рис. 4.13. Досліджуваний розчин вносять в електролітичну комірку (електролізер), де роль анода виконує ртуть, що знаходиться на дні електролізера. Катодом є ртутний краплинний електрод, сполучений з резервуаром ртуті. Через електролізер проходить струм, напругу якого можна плавно змінювати реохордом. Сила струму вимірюється гальванометром.
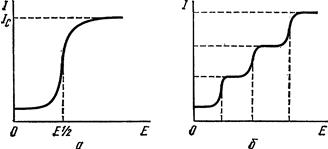
Рис. 4.14. Полярографічні криві: а – для моно-компонентного розчину; б – для полікомпо-нентного розчину. У момент утворення і відриву краплі ртуті напруга автоматично змінюється і залежність струму від напруги при цьому записується графічно. Типова поляризаційна крива наведена на рис. 4.14. При збільшенні напруги до величини потенціалу розкладу, при якому відбувається відновлення на катоді катіонів розчину, струм у колі різко зростає і досягає максимальної величини. Характеристикою досліджуваної речовини (катіона) є потенціал півхвилі ½Е, тому що потенціал початку її визначити важко. Концентрація відновленої речовини в розчині може бути визначена за величиною граничного дифузійного струму. Залежність струму і від прикладеної напруги Е при оборотному електродному процесі виражається рівнянням оборотної полярографічної хвилі:
де: Е½ – потенціал півхвилі; id – дифузний струм. У випадку необоротних процесів рівняння полярографічної хвилі ускладнюється. При і = ½іd рівняння (4.36) перетвориться на: Е = Е½, (4.37) Отже, потенціалом півхвилі називають таку величину потенціалу, при якій струм зростає до половини граничного значення. Якщо в розчині є декілька речовин, здатних до відновлення на ртутному катоді, то на полярограмі буде кілька хвиль (рис. 4.14 б). Аналізуючи полярограми, можна встановити якісний склад розчину і концентрацію розчинених речовин. Цей метод має високу чутливість (10–4–10–7 моль/л) і дає можливість досліджувати мінімальні дози (0,2–0,3 мл) сумішей розчину речовин.
КОНТРОЛЬНІ ЗАПИТАННЯ 1. Що таке питома електропровідність? 2. Поняття про еквівалентну електропровідність. 3. Сформулювати закон Кольрауша і записати формулу, яка випливає з цього закону. 4. Як виміряти опір провідників другого роду? 5. Як визначити точку еквівалентності при кондуктометричному титруванні? 6. Чому електропровідність розчинів визначають за допомогою змінного струму? 7. Як побудована шкала електродних потенціалів? 8. Що таке електроди першого роду? 9. Що таке електроди другого роду? 10. Що таке нормальний електродний потенціал і як його визначити? 11. Пояснити суть методу компенсації для вимірювання е.р.с. 12. Пояснити суть дифузійного і мембранного потенціалів, їх величину та значення в біології. 13. Пояснити принцип дії гальванічного елемента Якобі-Даніеля. 14. Написати формулу рівняння Нернста. Які показники можна визначити, використовуючи рівняння Нернста? 15. Пояснити принцип дії концентраційного елемента. 16. Охарактеризувати будову і принципи дії водневого електрода. 17. Принцип роботи хлорсрібного електрода. 18. Поняття про нормальний редокс-потенціал. 19. Біологічне значення редокс-потенціалів. 20. Принцип роботи каломельного електрода. 21. У чому полягає суть визначення рН за допомогою скляного електрода? 22. Принцип роботи полярографа. Поняття про потенціал півхвилі. 23. Переваги методу полярографії.
Розділ V ХІМІЧНА КІНЕТИКА І КАТАЛІЗ Хімічна кінетика (від грецьк. kinetikos – приводити в рух) – це вчення про швидкість хімічних реакцій і її залежність від різних факторів (природи, концентрації реагуючих речовин, температури, тиску, каталізаторів та ін.). Вивчення кінетики і механізму хімічних реакцій має велике теоретичне і практичне значення як у хімії, так і в біології. Хімічні реакції можуть відбуватися з різними швидкостями. Деякі реакції, супроводжуючись вибухом, закінчуються за тисячні частки секунди, інші ж здійснюються протягом хвилини, годин і навіть багатьох років. Хімічна кінетика складає основу важливих напрямків сучасної хімії і хімічної промисловості. За допомогою її законів розробляють раціональні принципи управління хімічними реакціями, стимулюють корисні і пригнічують небажані процеси. Хімічна кінетика є фундаментом для вивчення біохімії, клінічної біохімії та інших біологічних наук. Для фізичної хімії найбільш важливим є вивчення кінетики каталізу, де каталізаторами виступають речовини білкової природи – ферменти (від лат. fermentum – закваска). Обмін речовин на клітинному рівні регулюється швидкістю синтезу, концентрацією ферментів, здійснюваною на генетичному рівні. Закони хімічної кінетики використовуються для пояснення ряду фізичних процесів (радіоактивний розпад), реакцій і явищ (нормальний і злоякісний ріст тканини, внаслідок радіоактивного опромінення). Знання закономірностей хімічної кінетики дає змогу обирати відповідні умови для проведення утилізації відходів промислових підприємств, контролювати забруднення повітря, водосховищ. Таким чином, вивчення основних законів хімічної кінетики відкриває шлях до покращення екологічного стану довкілля.
§1. Основні поняття хімічної кінетики. Хімічна кінетика використовує спеціальні поняття і визначення. Хімічні реакції це перетворення одних речовин в інші, що можуть відрізнятися за хімічним складом і структурою у дуже широких межах. Вони характеризуються стехіометричним співвідношенням речовин, які беруть у них участь, ступенем перетворення, константами швидкості та рівноваги, енергією активації і тепловим ефектом. Такі реакції класифікуються за типом фазового стану реагуючої системи (газо-, рідинно- і твердофазові), типом частинок (йонні, радикальні, молекулярні), числом молекул, що беруть участь у процесі (моно-, ди- і полімолекулярні), кінетичним механізмом (послідовні, паралельні, спряжені), характером хімічного процесу (розкладу, полімеризації, окиснення, приєднання). Крім того, розрізняють гомо- і гетерогенні реакції. Перші з них відбуваються в усьому об’ємі фази, другі – на межі фаз. Речовини, які вступають у хімічну реакцію, називають реагентами, а хімічні сполуки, утворені внаслідок реакції – продуктами. Крім того, в багатьох випадках, особливо у живих організмах, на різних етапах хімічної реакції утворюються речовини, які не входять до кінцевих продуктів. Ці речовини називаються проміжними сполуками (метаболітами), а реакції, в яких виникають і використовуються проміжні сполуки – проміжними реакціями. Сукупність стадій, із яких складається хімічна реакція, зі структурними змінами рагуючих речовин складає механізм реакції.
§2. Залежність швидкості хімічної реакції від концентрацій реагуючих речовин. В основі хімічної кінетики лежить швидкість хімічної реакції, яка вимірюється змінами концентрації реагуючих речовин за одиницю часу при постійному об’ємі реакційної суміші. Молекули тієї чи іншої системи можуть взаємодіяти тільки при зіткненні. Реакція відбувається швидко при великому числі зіткнень, яке залежить від концентрацій реагуючих речовин. Згідно з законом діючих мас (К.Гульдберг і П.Вааге, 1867), швидкість хімічної реакції у газовому середовищі або розчині у кожний окремий момент пропорційна добутку концентрацій реагуючих речовин. Як приклад можна розглянути реакцію, у якій всі речовини перебувають у газоподібному стані: А+В = С+D. Чим вища концентрація молекул А і В, тим більше загальне число їх ефективних зіткнень, що супроводжується утворенням нових речовин С і D. Для такої реакції швидкість взаємодії між молекулами А і В виразиться рівнянням: υ = kСАСВ, де: k – коефіцієнт пропорційності, який називається константою швидкості хімічної реакції; СА і СВ – концентрації реагуючих речовин. Константа швидкості хімічної реакції для кожної окремої реакції при постійній температурі є величина стала. З використанням у реакції вихідних речовин швидкість буде зменшуватися. Середня швидкість реакції υ за даний проміжок часу τ2–τ1 ,буде
де ΔС – зміна концентрації реагуючої речовини за проміжок часу Δτ. Середня швидкість реакції υ завжди вважається величиною додатною. Відношення у правій частині рівняння може бути як додатним, так і від’ємним залежно від того, яким методом вивчають кінетику процесу: за зміною концентрації вихідних речовин чи за накопиченням продуктів реакції. У першому випадку С2<С1 тому, щоб середня швидкість мала додатнє значення. Праву частину беруть зі знаком “–” (рис. 5.1, крива СА). Навпаки, у другому випадку, коли С2>С1, то праву частину рівняння беруть зі знаком “+” (рис. 5.1, крива СВ). Однак, протягом хімічного процесу проходить зміна концентрації реагуючих речовин, причому концентрація продуктів реакції збільшується, а вихідних речовин зменшується. Отже, в різний момент часу швидкість хімічної реакції є різною.
Рис. 5.1. Зміна концент-рації реагенту СА і про-дукту СВ з часом реак-ції. Якщо віднести зміну концентрації речовини до нескінченного малого проміжку часу, то можна визначити істинну швидкість реакції.
Таким чином, впродовж реакції швидкість її змінюється, оскільки змінюються концентрації реагуючих речовин і продуктів реакції. Так, наприклад, за 5 хв концентрація продукту, що утворюється в процесі реакції, змінюється від 0,10 до 0,30 моль/л. Тоді швидкість реакції дорівнює:
Усі висновки щодо залежності швидкості реакції від концентрації реагуючих речовин не поширюються на тверді речовини. Коли в реакції з газами або рідинами беруть участь тверді речовини (гетерогенні системи), то в реакцію вступають лише ті молекули твердої речовини, що є на її поверхні. Тому при взаємодії твердої речовини з рідкою (наприклад, металу з кислотою), або твердої речовини з газоподібною (горіння вугілля) швидкість реакції залежить не тільки від концентрації кислоти чи кисню, а й від величини поверхні S твердих реагентів: V = kSC = k’C, (5.2) де k’ – константа швидкості з урахуванням величини поверхні твердого реагенту. Роль поверхні в гетерогенних реакціях значна, і збільшення її спричинює збільшення швидкості реакції. Так, подрібнене вугілля, має велику сумарну площу поверхні і згоряє швидше, ніж вугілля у великих кусках. Важливим чинником, що впливає на швидкість гетерогенної реакції, є також дифузія, тобто надходження до поверхні твердого реагенту нових порцій реагуючих речовин. Штучне прискорення процесу дифузії струшуванням, пермішуванням веде до зростання швидкості реакції.
§3. Залежність швидкості хімічної реакції від температури. Енергія активації хімічних реакцій. Залежність реакції від температури визначається правилом Вант-Гоффа, згідно з яким з підвищенням температури на кожні 10 градусів швидкість більшості реакцій збільшується в 2-4 рази. Математично ця залежність виражається рівнянням:
де υ1 і υ2 – швидкості реакцій відповідно при температурах τ1 і τ2, а γ – температурний коефіцієнт реакції. Він показує, у скільки разів збільшується швидкість реакції з підвищенням температури реакційної суміші на 10ºС. Наприклад, якщо збільшити температуру від 10 до 40ºС, при γ = 3, швидкість реакції збільшується в 27 разів:
В організмі людини і тварин реакції відбуваються під впливом білкових каталізаторів – ферментів. Зі збільшенням температури швидкість біохімічних реакцій відповідно зростає. При високих температурах (50-60ºС) каталітичні властивості ферментів втрачаються внаслідок термоденатурації білка і швидкість реакції різко зменшується. Отже, для хімічних процесів, що проходять в організмі, виявляється так званий температурний оптимум, який для теплокровних лежить в інтервалі 36-42ºС. При вивченні закономірності проходження реакції важливе значення має енергія активації. Швидкість будь-якої хімічної реакції залежить від числа зіткнень між молекулами реагуючих речовин за одиницю часу. Однак, не всі зіткнення ведуть до утворення продуктів реакції. Так, якщо середня швидкість молекули газу при 0ºС і тиску 1 атм дорівнює 105 см/сек, а довжина вільного шляху (між зіткненнями молекул) рівна 10–5 см, то число зіткнень для одної молекули буде відповідно 1010 за 1сек. Однак, швидкість багатьох газових реакцій дуже мала. Це пояснюється тим, що далеко не кожне зіткнення молекул може призвести до взаємодії між ними. Наприклад, в реакції 2HJ → J2+H2 із величезного числа зіткнень молекул (2·1017) при тиску 1 атм і 556°С тільки одне зіткнення є ефективним і супроводжується хімічною реакцією. Число ефективних зіткнень, порівняно з числом реальних зіткнень, як правило, дуже мале, що може бути відображено в рівнянні:
або
де: k – константа швидкості; А – постійний множник; е – основа натуральних логарифмів; Е – енергія активації; Т – абсолютна температура; R – універсальна газова стала. Згідно з теорією активації Арреніуса (1889), при зіткненнях у взаємодію вступають тільки ті молекули, які володіють відповідним запасом енергії, необхідної для здійснення тої чи іншої реакції. Ці молекули називаються активними молекулами і від їх концентрації залежить швидкість реакції. Кількісне число активних молекул можна вирахувати із закону розподілу Максвелла-Больцмана (1859):
де: N – кількість активних молекул; N0 – загальне число молекул. При сталій температурі число активних молекул залишається постійним. Енергією активації називається та мінімальна надлишкова енергія, яку потрібно надати молекулам, щоб між ними відбулась хімічна взаємодія. Цю енергію виражають в кДж/моль. Величина енергії активації може бути визначена розрахунковим методом із залежності константи швидкості від температури, яку одержано експериментальним шляхом. Для цього використовують рівняння 5.5, яке при переході до десяткових логарифмів матиме вигляд:
Більшого розповсюдження набув метод визначення енергії активації шляхом вимірювання швидкості реакції при двох різних температурах; так, наприклад, використовуючи рівняння (5.4), при Т1 і Т2 відповідно для k1 і k2:
Якщо поділити першу величину на другу, то постійний множник (А) вилучається:
або
звідки можна визначити величину енергії активації:
§4. Методи визначення швидкості хімічних реакцій. Кінетична класифікація хімічних реакцій. Швидкість хімічних реакцій вимірюється як швидкість зменшення концентрації вихідних речовин або як швидкість збільшення концентрації продуктів реакції. Методи визначення швидкості хімічних реакцій поділяють на дві групи – диференційні і інтегральні. Інтегральні застосовуються частіше. Вони базуються на використанні фізичного (поляриметрія, газова хроматографія, спектроскопія, мас-спектроскопія, манометрія) і хімічного (титрування) аналізів. Усі хімічні реакції класифікуються за ознакою молекулярності реакції, порядку реакції, а окремі види складних хімічних реакцій поділяють на паралельні, послідовні, спряжені, оборотні, ланцюгові. Молекулярність реакції визначається числом молекул, які вступають у хімічну взаємодію в елементарному акті реакції. Елементарний акт хімічної реакції – це такий акт хімічного перетворення, який відбувається за участю найменшої кількості реагуючих частинок. За ознакою молекулярності реакції поділяються на мономолекулярні, бімолекулярні (двомолекулярні) і тримолекулярні. Тримолекулярні зустрічаються рідко. Мономолекулярними називають реакції, в елементарному акті яких бере участь тільки одна молекула. До таких реакцій належать реакції розкладу, ізомеризації, дисоціації, радіоактивний розпад. Прикладом може бути розщеплення фруктозо-1,6-дифосфату альдолазою до двох фосфотріоз у гліколізі:
Бімолекулярні реакції – це реакції, елементарний акт яких здійснюється при зіткненні двох молекул: А+В → АВ. До такого типу реакцій належать реакції сполучення та обміну. Приклад такої реакції – утворення гідроген хлориду: Н2+Сl2 → 2HCl. Тримолекулярні реакції – це реакції, елементарний акт яких здійснюється при зіткненні трьох молекул. Прикладом може бути реакція горіння карбон (ІІ) оксиду: 2СО+О2 → 2СО2. Для характеристики кінетики хімічних процесів, які вивчаються експериментально, введено поняття порядок реакції, яке принципово відмінне від поняття молекулярність. Порядок хімічної реакції визначається за більш формальною ознакою, ніж її молекулярність – за видом рівняння, що виражає залежність швидкості реакції від концентрації реагуючих речовин. Розрізняють реакції першого (υ = KC), другого (υ = KC2), третього (υ = KC3), а також нульового порядків. Порядок реакції за даною речовиною – це число, що дорівнює показнику ступеня, в якому концентрація цієї речовини входить у кінетичне рівняння реакції: υ =KCаАСвВ, (5.12) де: υ – швидкість хімічної реакції; СА і СВ – концентрації реагуючих речовин; а та в – їх стехіометричні коефіцієнти. Порядок за речовиною А дорівнює а, за речовиною В – в. Загальний порядок реакції визначається сумою показників ступеня (а+в), з якими концентрації всіх вихідних реагентів входять до кінетичного рівняння швидкості хімічної реакції (5.12). Наприклад, швидкість реакції утворення гідроген йодиду в пароподібній фазі H2+J2 → 2HJ дорівнює 2HJ → H2+J2 також другий, тому що кінетичне рівняння цієї реакції: Реакції, для яких в досліді спостерігається перший порядок реакції, але в той же час вони є бімолекулярними, називаються псевдомолекулярними. Реакції на поверхні часто проходять за нульовим порядком. Наприклад, швидкість розкладу амоніаку на вольфрамі при 1138 К і тиску, більшому від 0,29·105 Па, або швидкість розкладу гідроген йодиду на золоті не залежить від концентрації реагентів в об’ємі системи. У цьому випадку реакції відбуваються за нульовим порядком, тобто їх швидкість не залежить від концентрації (парціального тиску) амоніаку або гідроген йодиду. У живих організмах реакції нульового порядку характерні для початкових стадій ферментативного каталізу, тобто при високому насиченні субстратом фермента. За своєю кінетичною характеристикою вони належать до моно- і бімолекулярних реакцій. Усі хімічні реакції поділяють на прості і складні. До перших належать хімічні реакції, які відбуваються лише в одному напрямку і в одну стадію: С+О2 → СО2. Складні реакції поділяються на паралельні, послідовні, оборотні, спряжені, ланцюгові. Паралельними називають такі реакції, при яких з одних і тих же вихідних речовин утворюються різні продукти:
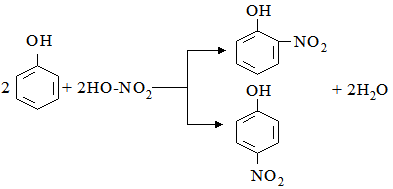
При нітруванні фенолу нітратною кислотою одночасно можуть утворюватися о- і n-нітрофеноли:
Послідовні реакції – це такі хімічні реакції, в яких утворення кінцевого продукту з вихідних речовин відбувається через декілька проміжних стадій. Прикладом може бути гідроліз крохмалю: (С6Н10О5)n → (С6Н10О5)х → mС12Н22О11 → nC6H12O6. крохмаль декстрини мальтоза глюкоза Оборотними називають такі реакції, при яких кінцеві продукти реакції, взаємодіючи між собою, утворюють вихідні речовини: аА+вВ ↔ сС+dD. Швидкість цих реакцій дорівнює різниці між швидкостями прямої і оборотної реакцій: А+В Якщо швидкість прямого процесу υ1 буде дорівнювати швидкості оборотного процесу υ2, то такий стан системи називається станом хімічної рівноваги. Якщо константу швидкості прямої реакції позначити k1, оборотної – k2, швидкість прямої – υ1, оборотної – υ2, концентрації речовин А–С1, В–С2, С–С1’, D–С2’, то швидкість прямої реакції буде: υ1 = k1С1С2. Швидкість оборотної реакції виражатиметься рівнянням: υ2 = k2С1’С2’. У стані хімічної рівноваги, коли:
|
||||
|
|
Последнее изменение этой страницы: 2024-06-27; просмотров: 8; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.135.188.211 (0.015 с.) |

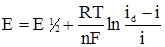 , (4.36)
, (4.36)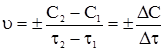 , (5.1)
, (5.1)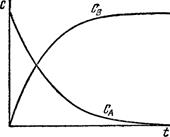
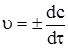 у даний момент часу.
у даний момент часу.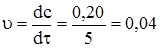 моль/л·хв.
моль/л·хв.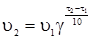 або
або 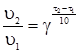 , (5.3)
, (5.3)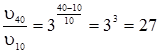 .
.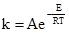 , (5.4)
, (5.4)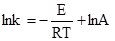 , (5.5)
, (5.5)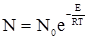 , (5.6)
, (5.6)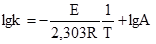 . (5.7)
. (5.7)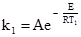 і
і 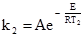 . (5.8)
. (5.8)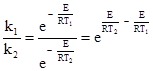 , (5.9)
, (5.9)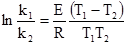 , (5.10)
, (5.10)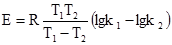 . (5.11)
. (5.11)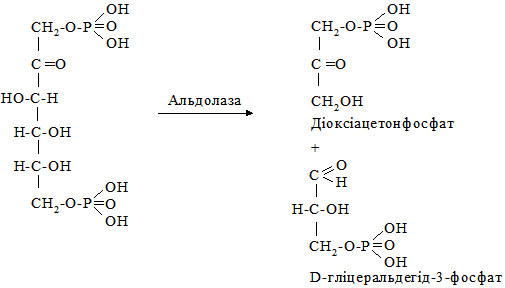
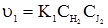 . Загальний порядок цієї реакції – другий, за воднем та йодом – перший. Порядок обортної реакції
. Загальний порядок цієї реакції – другий, за воднем та йодом – перший. Порядок обортної реакції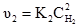 . Таким чином, реакції, в рівняння швидкості яких входить концентрація реагуючої речовини в першому ступені, називаються реакціями першого порядку, а реакції, швидкість яких пропорційна добутку двох концентрацій або квадрату концентрації, називаються реакціями другого порядку.
. Таким чином, реакції, в рівняння швидкості яких входить концентрація реагуючої речовини в першому ступені, називаються реакціями першого порядку, а реакції, швидкість яких пропорційна добутку двох концентрацій або квадрату концентрації, називаються реакціями другого порядку.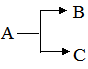
 .
.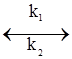 С+D.
С+D.


