Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Эритремия (истинная полицитемия)Содержание книги
Поиск на нашем сайте
Глава V БОЛЕЗНИ СИСТЕМЫ КРОВИ
Гемобластозы............................................................................ 404 Острый лейкоз.......................................................................... 406 Хронические лейкозы................................................................. 416 Хронический миелолейкоз......................................................... 417 Эритремия (истинная полицитемия)...................................... 423 Хронический лимфолейкоз........................................................ 430 Множественная миелома......................................................... 435 Анемии......................................................................................... 443 Железодефицитная анемия...................................................... 443 Сидероахрестическая анемия.................................................. 449 В\2-дефицитная анемия............................................................ 450 Гемолитические анемии........................................................... 455 Наследственный микросфероцитоз........................................ 457 Талассемия................................................................................. 458 Аутоиммунная гемолитическая анемия.................................. 460 Пароксизмальная ночная гемоглобинурия............................... 463 Гипопластические (апластические) анемии........................... 465 Геморрагические диатезы........................................................ 468 Тромбоцитопенические пурпуры............................................. 472 Идиопатическая тромбоцитопеническая пурпура............... 473 Гемофилии.................................................................................. 475 Ангиопатии (вазопатии)........................................................... 477 Наследственная геморрагическая телеангиэктазия (синдром Ослера — Рандю)...................................................... 478 Диссеминированное внутрисосудистое свертывание крови . 479 ГЕМОБЛАСТОЗЫ Гемобластозыпредставляют собой опухолевые заболевания кроветворной ткани. Их подразделяют на две большие группы — лейкозы и гематосаркомы. Лейкозы — опухоли из кроветворной ткани с первичной локализацией в костном мозге. Опухолевые клетки легко выходят в периферическую кровь, давая характерную гематологическую картину. Гематосаркомы — опухоли из кроветворной ткани с первичной внекостномозговой локализацией и выраженным местным опухолевым ростом. Лейкозы и гематосаркомы связаны между собой генетическим родством клеток, из которых они происходят, и могут переходить друг в друга. Все лейкозы делят на острые и хронические на основании не клинической характеристики, а морфологических особенностей опухолевых клеток, составляющих субстрат лейкоза. При острых лейкозах субстратом опухоли являются так называемые властные клетки, при хронических — созревающие и зрелые клетки.
Разделение острых лейкозов на отдельные формы основывается на цитохимических особенностях бластных клеток, в которых с помощью цитохимических исследований выявляют определенные вещества и ферменты. Хронические лейкозы с достаточной уверенностью разделяют на различные формы на основе морфологических свойств клеток, составляющих опухоль. Выделяют хронические лейкозы миелопролиферативной и лимфопролиферативной природы; подобное разделение основано на том, какая клетка-предшественница — миело- или лимфопоэ-за, подвергается мутации. Гематосаркомы в зависимости от типа клеток, составляющих опухоль, делят на лимфогранулематоз (с обязательным присутствием клеток Березовского — Штернберга и Ходжкина) и так называемые нелимфогрануле-матозные (неходжкинские) лимфомы. Среди нелимфогранулематозных лимфом в зависимости от морфологии опухолевых клеток выделяют лим-фоцитарные (зрелоклеточные), лимфобластные (малодифференцирован-ные) и гистиоцитарные (ретикулосаркомы). Этиология.Причины гемобластозов окончательно неясны. Мужчины болеют чаще, чем женщины (3:2), у детей и лиц пожилого и старческого возраста гемобластозы встречаются чаще, чем у лиц зрелого возраста. Установлен ряд факторов, влияющих на частоту заболевания гемо-бластозами. • Радиационный: ионизирующая радиация в атмосфере, облучение • Химический: бензол и другие токсичные вещества, в том числе ле • Наследственные хромосомные дефекты. Обменные нарушения — изменение обмена триптофана (лейкозоген-ное действие метаболитов триптофана).
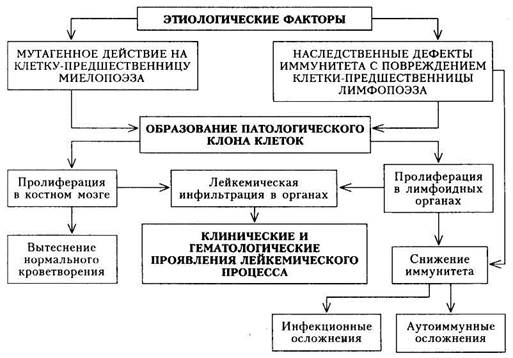
Схема 23. Патогенез лейкоза Вирусный фактор (в частности, вирус Эпштейна— Барр). Все указанные факторы имеют значение при определенных формах гемобластозов, однако несомненное их участие в происхождении заболевания в каждом конкретном случае не установлено.
Патогенез.Предполагают, что этиологический фактор повреждает ДНК кроветворной клетки, нарушает генетический код и приводит к безостановочному размножению и нарушению дифференциации той или иной разновидности клеток. В соответствии с этой точкой зрения в настоящее время общепризнанной является клоновая теория патогенеза гемобластозов, как и опухолей вообще. Лейкозные клетки представляют собой клон, потомство одной мутировавшей клетки. Полагают, что мутация происходит почти непрерывно, однако мутировавшие клетки уничтожаются системой фагоцитирующих макрофагов или иммунными силами организма. Можно предполагать, что для развития опухоли, в том числе и гемоблас-тоза, необходимо сочетание мутации клеток и ослабления иммунной защиты. Дальнейшее распространение опухоли осуществляется путем метаста-зирования этих клеток по кроветворной системе. Предполагается также, что гемобластозы происходят из клеток 1-го и 2-го классов схемы кроветворения. Иначе говоря, родоначальником опухолевого процесса кроветворной ткани чаще всего является клетка-предшественница миелопоэза или лимфопоэза. Другой патогенетической особенностью многих гемобластозов является постепенное озлокачествление опухолевого процесса, обозначаемое термином «опухолевая прогрессия». Опухолевая прогрессия выражается сле- дующими процессами: 1) угнетением нормального кроветворения; 2) наступлением «бластного криза» (смена дифференцированных опухолевых клеток недифференцированными); 3) появлением способности лейкозных клеток расти вне органов кроветворения; 4) уходом лейкозных клеток из-под контроля цитостатических препаратов; 5) неодинаковыми свойствами лейкозных клеток в разных очагах лейкозной пролиферации. Эти проявления опухолевой прогрессии находят свое клиническое выражение при гемобластозах (схема 23). ОСТРЫЙ ЛЕЙКОЗ Острый лейкоз (ОЛ) — опухолевое заболевание кроветворной системы, при котором субстрат опухоли составляют бластные клетки. Опухолевая трансформация осуществляется на самых ранних этапах дифференцирования стволовой кроветворной клетки и дальнейшего ее созревания не происходит. Термин «острый лейкоз» отражает не временной фактор (длительность течения болезни), а морфологические и цитохимические особенности опухолевых клеток. ОЛ — одно из наиболее тяжелых заболеваний из группы гемобласто-зов, они занимают основное место в структуре заболеваемости этими процессами, составляя приблизительно V3 от их общего числа. Встречаются О Л у лиц любого возраста, при этом отмечается два пика заболеваемости: в возрасте 3 — 4 и 60 — 69 лет (мужчины болеют чаще, чем женщины). В прошлом (до середины 60-х годов) ОЛ в течение нескольких месяцев приводил к смерти больных (на этом основании ранее был предложен термин «острый лейкоз»). В настоящее время достигнуты значительные успехи в его лечении: в большинстве случаев (20 — 40 %) удается достигнуть полной ремиссии, которая может длиться более 5 лет у 10 — 20 % больных острыми нелимфобластными лейкозами и у 50 % больных острыми лимфо-бластными лейкозами. В успехе лечения ОЛ исключительно большое значение имеет фактор времени: чем раньше будет диагностировано заболевание и чем раньше будет начато лечение, тем больше шансов на его успех.
Этиология.Этиология заболевания неизвестна. Тем не менее признается существование ряда факторов, способствующих развитию ОЛ: 1) хромосомные аномалии; 2) облучение; 3) токсические влияния вследствие загрязнения внешней среды, лекарственной терапии; 4) предшествующие заболевания системы кроветворения (рефрактерные формы анемий, пароксиз-мальная ночная гемоглобинурия, так называемые миелодисплазии). Эпидемиологические исследования показывают, что в семьях больных О Л риск заболеваемости повышается почти в 3 — 4 раза. На значение генетических факторов в развитии ОЛ указывает и увеличение заболеваемости при некоторых генетических нарушениях и аномалиях развития (болезнь Дауна, некоторые виды анемий и др.). Предполагается, что роль генетических факторов ограничивается формированием предрасположенности к развитию ОЛ, которая затем реализуется при воздействии различных факторов (лучевых, химических, экзо- и эндотоксических воздействий). Патогенез.Как уже говорилось, в основе болезни лежит пролиферация опухолевых бластных клеток в костном мозге, которые затем метаста-зируют в различные органы (селезенка, печень, головной мозг и пр.). ОЛ сопровождается угнетением нормального ростка кроветворения, что связа- но с несколькими факторами: 1) «повреждение» кроветворного окружения вокруг опухоли; 2) «вытеснение» нормального ростка гемопоэза; 3) выработка властными клетками ингибиторов, подавляющих рост нормальных кроветворных клеток. Классификация.Опухолевая трансформация при ОЛ происходит на стадиях дифференцировки родоначальных кроветворных клеток. В связи с этим О Л в первую очередь разделяют на лимфобластные, т.е. относящиеся к клеткам-предшественницам лимфопоэза и составляющие 15 % всех ОЛ, и на нелимфобластные, относящиеся к клеткам-предшественницам миелопоэза и составляющие основную массу О Л у взрослых. Каждая из этих двух больших групп ОЛ неоднородна, и в них выделяют различные формы: 1) острый лимфобластный лейкоз (ОЛЛ): а) общая форма («ни Т ни В» форма) — до 70 % ОЛЛ; б) Т-форма - до 25 %; в) В-форма — до 3 — 5 %; 2) острый миелобластный лейкоз (ОМЛ) — до 60 %; 3) острый миеломонобластный лейкоз (ОММЛ) — до 20 %; 4) острый монобластный лейкоз (ОМнЛ) — 3 — 7 %; 5) острый промиелоцитарный лейкоз (ОПрЛ) — 2 — 5 %; 6) острый эритромиелоз (ОЭМ) — 2 — 5 %; 7) острый недифференцируемый лейкоз (ОНЛ) — 2 —3 %.
В течении ОЛ выделяется несколько стадий: I. Начальная оценивается чаще всего ретроспективно. II. Развернутая — с четкими клиническими и гематологическими про 1) первую «атаку»; 2) ремиссию (полную или неполную); 3) рецидив болезни; 4) второй рецидив и т.д. III. Терминальная — отсутствие эффекта от цитостатической тера Выделение стадий ОЛ необходимо прежде всего в практическом отношении, так как в различных стадиях проводится различное лечение (мощная цитостатическая терапия — в развернутой стадии в период первой «атаки» или рецидива, поддерживающая — в период ремиссии). Клиническая картина.Проявления ОЛ могут быть весьма многообразными, в связи с чем их можно представить в виде следующих «больших» синдромов: I. Гиперпластический синдром (называемый также синдромом лейке-мической пролиферации), обусловленный опухолевым ростом как в костном мозге, так и вне его (метастазирование): 1) увеличение селезенки, печени, лимфатических узлов (перифери 2) поражения кожи (кожные лейкозные инфильтраты — неспеци II. Анемический синдром. III. Геморрагический синдром — от мелкоточечных и мелкопятнис IV. Интоксикационный синдром — снижение массы тела, лихорадка, На I этапе диагностического поиска можно отметить ряд вариантов начала заболевания и дальнейшего его течения. Примерно в половине случаев (преимущественно у лиц молодого возраста) отмечается острое начало заболевания. Обычно это активные, энергичные люди, не привыкшие уделять внимание своему самочувствию и обращающиеся к врачу лишь в крайних случаях. Острое начало болезни протекает под видом ангины, гриппа, острого респираторного заболевания. У некоторых больных, помимо выраженной интоксикации и лихорадки, развивается тяжелый приступ болей в животе, сопровождающийся диспепсическими расстройствами. Таких больных нередко направляют в инфекционное отделение с подозрением на заболевания тифо-паратифозной группы. У 10 % больных заболевание начинается профузными кровотечениями (носовые, маточные, желудочно-кишечные); некоторые больные впервые обращаются по поводу гиперпластического гингивита и язвенного стоматита. Примерно в 20 % случаев начальная стадия ОЛ проходит мимо внимания врача и самого больного, что обусловлено неспецифическими симптомами, выраженными в весьма незначительной степени. Тем не менее при ретроспективной оценке развития болезни удается установить, что еще до обращения к врачу у больного отмечались нарастающая слабость, повышенная утомляемость, боли в костях, мышцах и суставах, незначительное увеличение лимфатических узлов или единичные кровоизлияния в кожу, на которые больные не обращали внимания, объясняя их мелкими травмами, и лишь при нарастании числа «синяков» обращались к врачу. Боли в костях и суставах иногда носят упорный характер, и такие больные могут находиться под наблюдением врача с диагнозом остеоар-троз, ревматоидный артрит (однако, к сожалению, анализ крови таким больным своевременно не производится).
Наконец, у 52 % больных явных изменений общего состояния не наступает, а заболевание обнаруживается при случайном исследовании крови (например, при диспансерном обследовании, оформлении санаторно-курортной карты и пр.). Более реальная возможность диагностики ОЛ возникает в развернутый период заболевания. Однако и в это время клиническая картина весьма вариабельна, так как те или иные синдромы выражены в разной степени. Синдром опухолевой интоксикации становится значительно выраженным и проявляется повышением температуры тела, резкой слабостью, потливостью, прогрессирующим снижением массы тела. Гиперпластический синдром проявляется увеличением лимфатических узлов и селезенки, обнаруживаемых самим больным. При метастатических поражениях жалобы весьма разнообразны: сильная головная боль, кашель, одышка, упорный «радикулит», боли в животе, рвота, понос, парестезии, кожный зуд — часто «уводят» мысль врача от ОЛ и дают возможность предполагать самостоятельные заболевания различных органов. При нарастании анемического синдрома признаки гипоксически-цир-куляторного синдрома начинают преобладать в клинической картине и дают основание предположить любую форму анемии. Точно так же проявления геморрагического синдрома могут быть весьма существенными (обширные геморрагии, кровотечения). Некоторые больные могут сообщить, что им раньше уже ставился диагноз О Л и проводилась цитостатическая терапия. В таком случае ухудшение состояния достаточно связать с ранее выявленным заболеванием. Таким образом, на I этапе диагностического поиска может быть достаточно данных для углубленного обследования больного (и обязательного проведения исследования крови). На II этапе диагностического поиска в развернутой стадии болезни можно обнаружить проявления указанных выше синдромов. Гиперпластический синдром проявляется: 1) увеличением лимфатических узлов, чаще шейных, с одной или с обеих сторон, плотноватой консистенции, безболезненных (характерно для ОЛЛ); 2) нерезко выраженным увеличением селезенки: плотноватая, безболезненная или слегка чувствительная, выступает из-под реберного края на 3 — 6 см); 3) увеличением печени: плотноватая, чувствительная, пальпируется на 2 — 4 см ниже реберного края. Эти физикальные данные необязательны при ОЛ и могут наблюдаться при других гемобластозах, лимфомах. Поражение других органов проявляется разнообразными симптомами. При поражении кожи обнаруживают плотноватые инфильтраты на коже розоватого или светло-коричневого цвета, обычно множественные (лейкемиды). В случае поражения легких наблюдаются симптомы бронхиальной обструкции (ослабление дыхания, удлинение выдоха, сухие хрипы), очаговые инфильтративные изменения (притупление перкуторного звука, ослабление дыхания или жесткое дыхание, сухие и влажные хрипы). Однако отличить специфический лейкозный пневмонит от бактериальной пневмонии, нередко осложняющей ОЛ, трудно. При поражении миокарда выявляют небольшое расширение границ сердца, тахикардию, глухие тоны сердца, в тяжелых случаях — признаки сердечной недостаточности. Поражение пищеварительного тракта при физикальном исследовании может проявляться болезненностью при пальпации в эпигастрии. При поражении ЦНС (нейролейкоз) обнаруживают ригидность затылочных мышц, симптом Кернига, нарушение функции черепных нервов, снижение мышечного тонуса и другие симптомы. Эти явления дают основание подозревать не ОЛ, а скорее самостоятельное заболевание того или иного органа. Анемический синдром проявляется бледностью кожных покровов, тахикардией, систолическим шумом во всех точках, снижением АД. Для геморрагического синдрома характерны кожные геморрагии петехиально-пятнистого характера, особенно выраженные при ОПрЛ. Следует иметь в виду, что указанные многообразные клинические проявления могут отсутствовать, и физикальное исследование в этих случаях не дает никакой информации. Поэтому предположение об О Л на II этапе может и не возникнуть. Решающим для диагноза О Л является III этап. Задачами III этапа диагностического поиска являются установление диагноза О Л и определение его формы, а также выявление органных поражений и осложнений. Этот этап диагностики относительно прост: для подтверждения опухолевой трансформации костномозгового кроветворения необходимо провести исследование периферической крови и костного мозга. При ОЛ клиническая симптоматика становится вполне определенной, когда «плацдарм» кроветворения существенно заменен бластными клетками. Считается, что в развернутой стадии болезни общая масса опухоли составляет 1012 клеток на 1 м2 площади тела. При исследовании периферической крови число лейкоцитов может колебаться от низких цифр до высоких — гиперлейкоцитоз (в настоящее время благодаря современной диагностике установлено, что оно составляет 20 —50109/л, весьма часто (в 50 % случаев) может наблюдаться лейкопения. Основным признаком ОЛ является наличие в крови опухолевых бластных клеток (бластемия). Эти клетки обнаруживаются в мазках крови в количестве от 5— 10 до 80 —90 %. Характерно так называемое лей-кемическое зияние — очень малое количество зрелых гранулоцитов — сег-ментоядерных и практически полное отсутствие палочкоядерных, юных, метамиелоцитов. При алейкемической фазе болезни бласты в периферической крови единичные или же вообще отсутствуют. В этих случаях диагноз ставят по результатам исследования пунктата костного мозга, при котором обнаруживается значительное повышение содержания бластных клеток (увеличение в стернальном пунктате бластных клеток более 30 % полностью подтверждает диагноз ОЛ). Существует правило: необходимо в течение всей жизни больного О Л хранить пунктаты костного мозга и мазки периферической крови, так как под влиянием проводимой цитостатической терапии картина крови и костного мозга становится нетипичной для О Л (в препаратах появляются так называемые терапевтические бласты с более грубым ядром и их практически нельзя отличить от лимфоцитов). При повторных обращениях больного в другие лечебные учреждения диагноз ОЛ может быть подтвержден лишь при изучении первичных препаратов. Другие гематологические показатели не имеют самостоятельного диагностического значения, хотя анемию и тромбоцитопению, сопутствующие ОЛ, нередко можно рассматривать как косвенные критерии диагноза ОЛ, особенно, если анемия и тромбоцитопения сочетаются с лихорадкой неясного генеза и спленомегалией. Анемия (обычно нормохромного и макроцитарного типа) усиливается по мере прогрессирования заболевания и рассматривается как проявление угнетения нормального кроветворения. То же происхождение имеет и тромбоцитопения. В некоторых случаях анемия и тромбоцитопения имеют аутоиммунное происхождение, что сопровождается умеренной желтушнос-тью, ретикулоцитозом, повышенным содержанием непрямого билирубина в крови, положительной пробой Кумбса. В некоторых случаях может быть так называемая малопроцентная форма ОЛ. Для нее характерны незначительно выраженная клиническая симптоматика, склонность к цитопении, особенно анемии, с увеличением бластных клеток до 20 % как в крови, так и в костном мозге. Эта картина может сохраняться несколько месяцев, в конечном итоге развивается типичная картина ОЛ. Властные клетки при всех формах ОЛ характеризуются крупными размерами, большим ядром, занимающим почти всю клетку и отличающимся нежно-сетчатым строением хроматина с крупными единичными ядрышками. Цитоплазма клеток в виде узкого ободка голубоватого или серо-голубого цвета, с единичными мелкими гранулами или без грануляции. Обычно морфологическое исследование окрашенных гематологическими красителями (в мазках крови) бластных клеток не позволяет с уверенностью дифференцировать формы ОЛ; исключение составляют бласты при ОПрЛ (обильная крупная фиолетовая зернистость цитоплазмы с наличием телец Ауэра и различная форма ядер клеток). Различать бластные клетки при разных ОЛ можно с большой достоверностью по их цитохимическим свойствам (табл. 14). Для определения ОЛЛ наиболее характерна PAS-реакция (окраска на гликоген), выявляющая в цитоплазме крупные гранулы, чаще в виде ожерелья. При ОНЛЛ в первую очередь характерна положительная реакция на пероксидазу, а для различных его форм, также в сочетании с положительной реакцией на неспецифическую эстеразу (ОМнЛ и ОММЛ), кислые мукополисахариды (ОПрЛ) и некоторые другие. Таблица 14. Цитохимическая характеристика бластных клеток при остром лейкозе
Форма ОЛ Цитохимические признаки бластных клеток
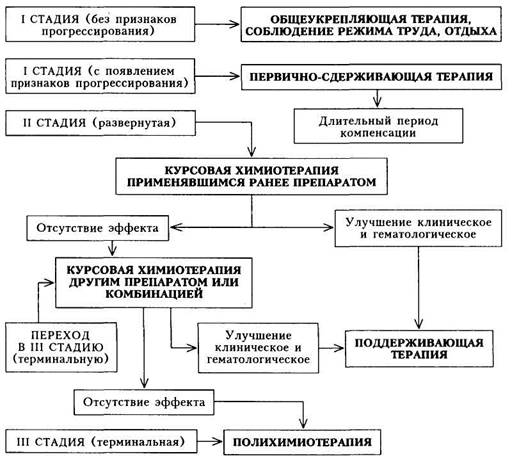
перокси-даза липиды гликоген (PAS-реакция) неспецифическая эстераза кислые мукополи-сахариды ОЛЛ омл ОММЛ ОМнЛ оэм онл 1 1 + + + 1 + (в отдельных клетках) ± + (гранулярно) + (диффузно) + (диффузно) ± (диффузно) + (гранулярно) 1 1 + + + 1 - Для дифференциации ОЛЛ большое значение имеют также иммунологические методы (обнаружение поверхностных иммуноглобулинов, способность к розеткообразованию и т.д.). Наконец, начинают приобретать все большую роль методы выявления поверхностных клеточных диффе-ренцировочных антигенов, которые в характерном для каждого типа клеток сочетании появляются и исчезают по мере созревания клетки (для этого используют специфические моновалентные антисыворотки). Определение степени органных поражений решается лабораторно-ин-струментальными методами и при необходимости — морфологическими исследованиями. Последние становятся особенно актуальными в периоде ремиссии для оценки ее полноты. С этой целью производят пункцию костного мозга увеличенных лимфатических узлов, селезенки, печени, инфильтратов кожи, увеличенного яичка. Обнаружение бластных клеток в пунктатах, а также в спинномозговой и плевральной жидкостях указывает на соответствующее органное поражение и исключает полную ремиссию после проведенной терапии. При подозрении на поражение лимфатических узлов средостения и корней легких проводят томографию и рентгенографию грудной клетки. В случае лейкозного пневмонита на рентгенограмме усилен легочный рисунок и выявляются мелко- и крупноочаговые тени по всей легочной ткани. При лейкемической инфильтрации миокарда могут быть изменения на ЭКГ в виде снижения вольтажа и появления отрицательных зубцов Т. Однако указанные изменения ЭКГ могут быть связаны не только со специфической инфильтрацией миокарда, но и с миокардиодистрофией, обусловленной опухолевой интоксикацией, кардиотоксическим действием цитостатических препаратов, применяемых при лечении ОЛ. При лейкемической инфильтрации печени ультразвуковое исследование выявляет ее увеличение и мелкоочаговые изменения эхогенности органа. Изотопная сцинтиграфия в подобных случаях также помогает обнаружить увеличение органа, снижение поглощения радиофармацевтического препарата с диффузно-неравномерным накоплением его. Однако эти изменения носят неспецифический характер и свидетельствуют лишь о диффузных изменениях ткани печени, которые могут развиться в результате неспецифического реактивного гепатита, а также токсического гепатита медикаментозного (цитостатики) генеза. В последнем случае правильной интерпретации указанных инструментальных данных будут способствовать результаты анамнеза, физикального исследования (нарастающее увеличение размеров печени, желтуха) и биохимических реакций, выявляющих значительную гиперферментемию, гипербилирубинемию. Специфическое поражение почек может проявляться при ультразвуковом исследовании увеличением органа, обычно двусторонним, с диффузными эхоструктурами в ткани. В анализе мочи — умеренная протеину-рия, гематурия, цилиндрурия. Эти изменения могут быть проявлением опухолевой интоксикации. Для нейролейкемии характерна картина спинномозговой жидкости: высокий цитоз, повышенное содержание белка и обнаружение в мазках бластных клеток. Серьезным осложнением цитоста-тической терапии является развитие агранулоцитоза, о котором судят по появлению в крови резкой лейкопении (до 1109/л и менее) и изменению лейкоцитарной формулы в виде уменьшения числа нейтрофилов (вплоть до исчезновения) и относительного увеличения количества лимфоцитов. Диагностика. Несмотря на то что клиническая картина ОЛ очерчена довольно ярко, все же признаков, патогномоничных только для этого заболевания, практически нет. В связи с этим даже такие, казалось бы, характерные для гемобластозов симптомы, как увеличение селезенки, печени, лимфатических узлов, анемия, позволяют врачу лишь заподозрить ОЛ и срочно провести исследование крови. Только морфологические методы — исследование костного мозга и периферической крови, позволяют диагностировать ОЛ. Существует ряд правил, которые следует помнить любому врачу, чтобы не пропустить ОЛ. Является обязательным динамическое исследование крови при всех рефрактерных к лечению и рецидивирующих ангинах, респираторных заболеваниях, гриппе, особенно если эти заболевания сопровождаются лимфаденопатией, геморрагическими проявлениями, а также артралгиями. Особая гематологическая настороженность должна быть при всех случаях лимфаденитов и гиперпластических гингивитов. Назначение таким больным различных физиотерапевтических и тепловых процедур без предварительного исследования крови может принести вред. Дифференциальная диагностика.При постановке диагноза ОЛ, в особенности при лейкопении, отсутствии гранулоцитов, следует проводить дифференциацию с рядом заболеваний. Агранулоцитозы не сопровождаются бластозом костного мозга, редукцией эритроидного роста кроветворения, геморрагический синдром отмечается редко, селезенка не увеличена. Гипопластические анемии не протекают с увеличением лимфатических узлов и селезенки. При стернальной пункции в мазке не выявляется увеличения количества бластных клеток. Важное значение для диагностики имеет трепанобиопсия: преобладание в трепанате жировой ткани свидетельствует о наличии гипопластической анемии. При диффузных заболеваниях соединительной ткани, хронических активных гепатитах могут наблюдаться лимфаденопатия, увеличение селезенки, анемия, тромбоцитопения, нейтропения, в пунктате костного мозга количество бластов может быть повышено до 10 — 20 %. В таких случаях необходима трепанобиопсия крыла подвздошной кости и повторные исследования крови и костного мозга. Кроме того, учет всей клинической картины помогает правильной оценке симптомов. С симптоматикой ОЛ сходна клиническая картина инфекционного мо-нонуклеоза - острое начало, наличие лихорадки, ангины; увеличение печени, селезенки, лимфатических узлов; лейкоцитоз с наличием мононук-леаров в гемограмме. Однако в отличие от О Л отсутствуют анемия и геморрагический синдром, увеличенные болезненные лимфатические узлы локализуются по заднему краю грудино-ключично-сосцевидной мышцы. В мазке крови вместо бластных клеток обнаруживают 60 70 % мононук-леаров, большинство из которых составляют средне- и широкоплазменные лимфоциты, увеличено количество моноцитов, тромбоцитопения отсутствует. Существенные трудности возникают при дифференциации ОЛ и хронического миелолейкоза (ХМЛ), дебютирующего бластным кризом. Клинические признаки, исследование крови и костного мозга не позволяют дифференцировать заболевания. В таких случаях существенную пользу оказывает исследование хромосом. Обнаружение Ph-хромосомы свидетельствует о хроническом миелолейкозе. Формулировка развернутого клинического диагнозаучитывает: 1) форму ОЛ; 2) стадию заболевания; 3) наличие внекостномозговых поражений; 4) наличие осложнений. Лечение.Цель современной терапии О Л у взрослых лиц — достижение длительной выживаемости (без возникновения рецидивов болезни). Это оказалось возможным при условии внедрения программного лечения, представляющего поэтапное уничтожение лейкозных клеток с помощью различных комбинаций цитостатических средств, а также комплекса мероприятий, направленных на борьбу (и предупреждение) с агранулоцито-зом, тромбоцитопенией, анемией, синдромом диссеминированного внутри-сосудистого свертывания крови (ДВС). Использование таких программ позволяет добиться ремиссии у 60 — 80 % взрослых больных и полного выздоровления от лейкоза у 20 — 30 %. При ОЛ руководствуются определенными принципами лечения. • Его начинают сразу же после постановки диагноза. • Оно должно быть дифференцированным и зависеть от морфологи • Одновременно назначают несколько препаратов (полихимиотера • Следует стремиться к достижению лейкопении. • В стадии ремиссии необходимо проводить поддерживающую тера Лекарственные препараты не способствуют превращению лейкемичес-ких клеток в нормальные, а уничтожают их. Основная задача терапии — санация организма от лейкемических клеток, что достигается применением большого количества химиопрепаратов с различным механизмом действия. Антилейкемические препараты наиболее активны по отношению к делящимся клеткам, причем некоторые из них активны в какой-то определенный период митоза (фазово- и циклоспецифические препараты — мер-каптопурин, цитобарин, или цитозар, метотрексат), другие — в течение всего митотического цикла (циклонеспецифические — циклофосфан, вин-кристин, преднизолон). Существуют определенные этапы при проведении химиотерапии: • вызывание ремиссии (индукция) проводится при первой «атаке» • закрепление (консолидация) ремиссии — 2 — 3 курса лечения пре • противорецидивная терапия (поддерживание ремиссии) проводится • профилактика нейролейкемии. Невыполнение одного из этапов такого программного лечения, необоснованные изменения в схемах химиотерапии приводят к неизбежному развитию рецидива и лишают больного шансов на полное выздоровление. Следует также отчетливо представлять, на основании каких признаков можно говорить о наступлении ремиссии или рецидива. Ремиссия характеризуется следующими показателями, сохраняющимися на протяжении не менее 1 мес: ▲в костном мозге число бластных клеток не превышает 5 %, пред ▲в периферической крови — абсолютное число нейтрофилов не ▲не имеется очагов экстрамедуллярного лейкемического роста Рецидив диагностируется при обнаружении в пунктате костного мозга 5 —20 % бластов (повторное обнаружение бластов в костном мозге более 5 % подтверждает рецидив), а также любого экстрамедуллярного лейкемического поражения (даже без вовлечения в патологический процесс костного мозга). При назначении полихимиотерапии следует иметь в виду, что те или иные морфологические варианты ОЛ оказываются чувствительными к комбинациям определенных препаратов. При остром лимфобластном и остром недифференцируемом лейкозах индукция ремиссии состоит из двух 4-недельных фаз: 1-я фаза: винкрис-тин, рубомицин, L-аспарагиназа, преднизолон; 2-я фаза: циклофосфан, цитозар, 6-меркаптопурин. Через 4 нед после завершения индукции проводится так называемая ранняя консолидация — цитозар в сочетании с вепезидом (2 пятидневных курса). В промежутках между пятидневными курсами назначают 6-меркаптопурин. Противорецидивная (поддерживающая) терапия проводится двумя цитостатическими препаратами — метотрексатом и 6-меркаптопурином. Профилактика нейролейкемии осуществляется с помощью эндолюм-бального введения метотрексата, цитозара и преднизолона или облучения головы с обоих латеральных полей. При остром нелимфобластном лейкозе индукция ремиссии проводится с помощью цитозин-арабинозида (цитозара) с рубомицином (курс длится 7 дней). Цитозар комбинируют также с митоксантроном; возможно сочетание рубомицина, цитозара и тиогуанина. Индукционная терапия предполагает проведение двух аналогичных курсов полихимиотерапии. Консолидация ремиссии осуществляется с помощью цитозара в комбинации с другими препаратами (митоксантрон). Поддерживающая терапия проводится цитозаром и рубомицином. Существуют и другие варианты поддерживающей терапии. На фоне поддерживающей терапии должны проводиться контрольные стернальные пункции (1 раз в 3 мес). Профилактика нейролейкемии осуществляется эндолюмбальным введением цитостатиков (метотрексат, цитозар) в сочетании с преднизолоном. Вместо этого осуществляют также облучение головы. При локализации очагов лейкемической инфильтрации в средостении, глотке, яичке проводится рентгенотерапия этих областей. В последние годы для лечения рецидива ОЛ или его профилактики применяют трансплантацию костного мозга (аллогенного или аутологич-ного, полученного в период ремиссии) после предварительного введения больших доз циклофосфана (50 мг на 1 кг массы тела в течение 4 дней) и однократного тотального облучения в дозе 10 Гр. Инфекционные осложнения ОЛ являются весьма грозными, в связи с чем активная терапия антибиотиками широкого спектра действия должна проводиться своевременно и в достаточных дозах. Профилактикой инфекционных осложнений, особенно у больных с гранулоцитопенией, является тщательный уход за кожей и слизистой оболочкой рта, помещение больных в специальные асептические палаты, стерилизация кишечника с помощью неадсорбируемых антибиотиков (канамицин, неоми-цин). При развитии геморрагического диатеза необходимо переливание тромбоцитной массы (1 — 2 раза в неделю) или свежей цельной крови. Прогноз.В процессе лечения могут быть достигнуты: 1) полная клинико-гематологическая ремиссия (клиническая компенсация без признаков лейкозной инфильтрации селезенки, печени и других органов, нормальный или близкий к норме анализ крови, в пунктате кост- ного мозга число бластных клеток не превышает 5 %, а общее число лим-фоидных и бластных клеток не более 40 %); 2) частичная клинико-гематологическая ремиссия (клиническая ком 3) выздоровление (состояние полной клинико-гематологической ре Ремиссии получают у 74 — 79 % взрослых больных, продолжительность ремиссий в среднем 2 года. Прогностически наименее эффективно лечение ОЭМ и всех форм ОЛ с парциальной цитопенией и панцитопе-нией. Профилактика.Первичной профилактики ОЛ не существует. Вторичная профилактика сводится к тщательному контролю за состоянием больного и правильному проведению противорецидивной терапии. Больных О Л ставят на диспансерный учет. ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ Как уже упоминалось, среди хронических лейкозов выделяют миело-пролиферативные и лимфопролиферативные заболевания. К числу хронических миелопролиферативных процессов относят хронический миелолей-коз, эритремию (истинную полицитемию), идиопатический миелофиброз (сублейкемический миелоз), эссенциальную тромбоцитемию (хронический мегакариоцитарный лейкоз, геморрагическая тромбоцитемия). Для всей группы миелопролиферативных заболеваний (лейкозов) характерен дефект (мутация) на уровне полипотентной стволовой клетки, этот дефект далее продуцируется на следующем классе стволовых клеток (олигопотентных) — клетках-предшественницах смешанной культуры. Эти клетки дают начало трем линиям миелоидного кроветворения — эрит-роцитарного, гранулоцитарного и мегакариоцитарного. Создается миело-идная пролиферация — основной признак, характеризующий субстрат этих заболеваний, при этом продукция клеток осуществляется из одного или нескольких ростков миелоидного кроветворения (эритробластическо-го, гранулоцитарного, мегакариоцитарного). Все сказанное объединяет, казалось бы, внешне различные заболевания миелопролиферативной природы — эритремию, хронический миелолейкоз, идиопатический миелофиброз, мегакариоцитарный лейкоз (тромбоцитемию). К числу хронических лимфопролиферативных заболеваний относят хронический лимфолейкоз (различные его формы), а также группу пара-протеинемических гемобластозов — заболеваний, при которых опухолевые клетки секретируют патологический белок (парапротеин); к ним относятся: множественная миелома (миеломная болезнь); болезнь Вальден-стрема (макроглобулинемия); болезнь тяжелых цепей. Далее будут рассмотрены наиболее часто встречающиеся хронические лейкозы: хронический миелолейкоз, эритремия, хронический лимфолейкоз, множественная миелома. ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ Хронический миелолейкоз (ХМЛ) — миелопролиферативное хроническое заболевание, при котором наблюдается повышенное образование гранулоцитов (преимущественно нейтрофилов, а также про-миелоцитов, миелоцитов, метамиелоцитов), являющихся субстратом опухоли. Этиология и патогенез.Причиной патологического роста клеток считается мутация клетки-предшественницы миелопоэза (частично детерминированная полипотентная клетка). Это доказывается обнаружением у больных ХМЛ специфического маркера — патологической Ph-хромосомы (филадельфийской) в клетках миелоидного, эритроидного, моноцитарно-го и тромбоцитарного рядов. Ph-хромосома является частым клеточным маркером, подтверждающим происхождение всего патологического клона клеток при ХМЛ от одной материнской. Несмотря на то что лейкозными являются все три ростка костного мозга, в развернутой стадии ХМЛ наблюдается безграничный рост, как правило, одного ростка — гранулоци-тарного. Существенно повышается в костном мозге продукция мегакарио-цитов, в периферической крови — тромбоцитов. По мере течения болезни моноклоновая стадия сменяется поликлоно-вой, что доказывается появлением клеток с различным неправильным набором хромосом. В этом проявляется закон опухолевой прогрессии, которому подчиняется данный лейкоз. ХМЛ чаще наблюдается у взрослых в возрасте 30 — 70 лет; отмечается небольшое преобладание мужчин. Классификация.Как отмечалось, заболевание закономерно проходит в своем развитии две стадии — моноклоновую и поликлоновую. Этому соответствуют три стадии хронического миелолейкоза в клиническом отображении: Стадия I — начальная — миелоидная пролиферация костного мозга + небольшие изменения в крови без явлений интоксикации. Стадия II — развернутая — выраженные клинико-гематологические проявления (интоксикация продуктами распада лейкозных клеток, увеличение печени и селезенки, миелоидная пролиферация костного мозга + изменения в крови). Стадия III — терминальная (соответствует развитию поликлоновой опухоли) — рефрактерность к проводимой цитостатической терапии, истощение, значительное увеличение селезенки и печени, дистрофические изменения внутренних органов, выраженные изменения крови (анемия, тромбоцитопения). Для терминальной стадии ХМЛ характерно развитие так называемых бластных кризов — появление в периферической крови бластных клеток (до 30 — 90 %), в связи с чем заболевание приобретает черты острого лейкоза. Чаще всего в костном мозге и периферической крови бластный криз характеризуется появлением миелобластов, однако могут встретиться и недифференцируемые бластные клетки. При кариологическом исследовании выявляется поликлоновость патологических клеток. Одновременно происходит значительное угнетение тромбоцитопоэза, развивается геморрагический синдром. Встречается также лимфобластный вариант бластно-го криза (в костном мозге и периферической крови появляется большое количество лимфобластов). 14-540 Клиническая картина.Клинические проявления ХМЛ могут выражаться большими синдромами: Миелопролиферативный синдром, в основе которого лежит миелоид-ная пролиферация костного мозга, включает: а) общие симптомы, вызванные интоксикацией, разрастаниями лей- б) увеличение печени и селезенки; в) лейкемические инфильтраты в коже; г) характерные изменения в костном мозге и периферической крови. Синдром, обусловленный осложнениями: а) геморрагический диатез (геморрагии и тромбозы вследствие нару б) гнойно-воспалительные (пневмонии, плевриты, бронхиты, гной в) мочекислый диатез (гиперурикемия вследствие повышенного рас Различная выраженность синдромов на разных стадиях болезни обусловливает достаточно полиморфную клиническую картину. Можно наблюдать больных, не предъявляющих никаких жалоб и вполне трудоспособных, и больных с тяжелыми поражениями внутренних органов, истощенных, полностью потерявших трудоспособность. На I этапе диагностического поиска в начальной стадии болезни больные могут не предъявлять жалоб, и заболевание будет диагностировано на последующих этапах. Жалобы общего характера (слабость, потливость, снижение массы тела) могут встречаться при самых разных заболеваниях, поэтому рассматривать их на I этапе как специфические для ХМЛ нельзя. Лишь позже, при выявлении других симптомов, указывающих на ХМЛ, они могут быть интерпретированы как выражение миелопролифе-ративного синдрома. Тяжесть и боли в области левого и правого подреберий обычно объясняются увеличением селезенки и печени. В сочетании с жалобами общего характера и болями в костях они могут ориентировать врача в отношении возможного объяснения этих симптомов миелопролиферативным синдромом. В терминальной стадии болезни часть жалоб может быть обусловлена возникновением осложнений: гнойно-воспалительных, геморрагического диатеза, мочекислого диатеза. На I этапе можно получить сведения об изменениях при исследовании крови и проводившемся ранее лечении (цитостатические препараты). Следовательно, если в поле зрения врача попадает больной, которому уже ставили диагноз ХМЛ, последующий диагностический поиск значительно упрощается. Важно выяснить у больных сведения о ранее проводившемся лечении и неэффективности препаратов, до данного момента улучшающих общее состояние, снижавших количество лейкоцитов. Такая информация заставит предположить переход в поликлоновую (терминальную) стадию болезни. На II этапе диагностического поиска возможно получение сведений, позволяющих высказать предположение: 1) о характере патологического процесса, т.е. существе самого заболевания; 2) о стадии заболевания; 3) о возможных осложнениях. В развернутой и терминальной стадиях выявляются признаки, в существенной мере подтверждающие предположение о ХМ Л: бледность кожных покровов (обусловлена нарастающей анемизацией), кожные геморрагии и инфильтраты (более характерны для терминальной стадии ХМЛ). Существенным признаком является спленомегалия (без увеличения лимфатических узлов), сочетающаяся с увеличением печени, что при соответствующих жалобах и анамнезе может быть расценено как проявление миелопролиферативного синдрома. При развитии осложнений, например инфаркте селезенки, отмечается резкая болезненность ее при пальпации, шум трения брюшины над селезенкой. Постепенно селезенка становится плотной (ее масса составляет 6 — 9 кг, спускается нижним полюсом в малый таз). Наиболее важные данные для диагноза ХМЛ получают на III э т а п е диагностического поиска. При исследовании периферической крови обнаруживают лейкоцитоз с появлением в лейкоцитарной формуле пролифе-рирующих форм (миелобласты и промиелоциты) и созревающих грануло-цитов (миелоциты, метамиелоциты); имеется базофильно-эозинофильная ассоциация. Число лейкоцитов колеблется в широких пределах, достигая в выраженных случаях 100 —200109/л, однако в терминальной стадии лейкоцитоз может значительно уменьшиться и даже развивается лейкопения. В ранних стадиях болезни возможно обнаружение гипертромбоцито-за. Развитие нормомакроцитарной анемии, связанной в основном с вытеснением лейкозным клоном красного ростка кроветворения, можно наблюдать в развернутой клинико-гематологической стадии. В терминальной стадии анемия становится еще более выраженной. При исследовании костного мозга обнаруживают миелоидную пролиферацию костного мозга, нормальный миелопоэз полностью замещен патологическим клоном. В мазке костного мозга преобладают гранулоциты: соотношение лейкоциты/эритроциты достигает 10:1, 20:1 за счет увеличения гранулоцитов. Если в периферической крови высокий тромбоцитоз, то в костном мозге отмечается большое количество мегакариоцитов. Функциональные свойства лейкоцитов и содержание в них ферментов изменены: снижена активность щелочной фосфатазы нейтрофилов, нарушена способность к фагоцитозу. При пункции увеличенной селезенки в развернутой стадии болезни обнаруживается преобладание миелоидных клеток. Данный этап оказывается решающим в идентификации бластного криза: нарастание количества бластных клеток в костном мозге и периферической крови (суммарное количество бластов и промиелоцитов равно 20 % и более, тогда как вне бластного криза это количество обычно не превышает 10-15 %). Сцинтиграфия костей помогает обнаружить увеличение плацдарма кроветворения (исследование производят при неясном диагнозе и оно не является обязательным для всех больных ХМЛ). Диагностика.Выявление ХМЛ в развернутой стадии болезни не представляет трудностей и основывается на характерных данных анализа крови, результатах исследования костного мозга, увеличении печени и селезенки. 14* Диагностическими критериями заболевания являются: • лейкоцитоз более 20109/л; • появление в лейкоцитарной формуле пролиферирующих форм (ми- • миелоидная пролиферация костного мозга (по данным миелограм- • снижение активности щелочной фосфатазы нейтрофилов (менее 25 • обнаружение Ph-хромосомы в кроветворных клетках; • расширение «плацдарма» кроветворения (по данным сцинтиграфии • увеличение размеров селезенки и печени. Дифференциальная диагностика.ХМЛ следует дифференцировать от так называемых лейкемоидных реакций, которые могут возникать при ряде заболеваний (туберкулез, рак, различные инфекции, почечная недостаточность и пр.). По определению А.И. Воробьева (1985), лейкемоид-ная реакция — это «изменения в крови и органах кроветворения, напоминающие лейкозы и другие опухоли кроветворной системы, но не трансформирующиеся в ту опухоль, на которую они похожи». При лейкемоид-ной реакции наблюдается высокий лейкоцитоз, в периферической крови появляются незрелые нейтрофилы, однако базофильно-эозинофильная ассоциация не обнаруживается. Дифференциальный диагноз основывается на выявлении основного заболевания (рак, туберкулез и пр.), а также на повышении активности щелочной фосфатазы нейтрофилов (вместо ее снижения при ХМЛ). При стернальной пункции для лейкемоидной реакции характерно увеличение содержания миелоцитов, однако Ph-хромосома никогда не определяется. Лечение.Основная задача лечения любого гемобластоза (в том числе и ХМЛ) — ликвидация или подавление роста патологического клона клеток. Однако применительно к хроническим лейкозам это не означает, что любого больного, у которого обнаруживается заболевание системы крови, сразу же нужно активно лечить цитостатическими препаратами, подавляющими опухолевый рост. • В начальной стадии болезни (при хорошем самочувствии, но несо • При появлении симптомов прогрессирования болезни необходимо
Схема 24. Принципы лечения хронического миелолейкоза тельном гематологическом контроле); после достижения клинической и(или) гематологической ремиссии решается вопрос о поддерживающей терапии. • В развернутой стадии болезни объем химиотерапии зависит от «группы риска», определяемой наличием неблагоприятных признаков: ♦ лейкоцитоз более 200109/л, бластемия более 3 %, сумма бластов ♦ снижение гемоглобина до уровня менее 90 г/л; ♦ тромбоцитоз более 500-109/л или тромбоцитопения менее ♦ сп леном егалия (селезенка пальпируется на 10 см ниже реберной ♦ гепатомегалия (печень пальпируется на 5 см ниже реберной дуги Низкий риск — наличие одного признака; промежуточный риск — наличие 2 — 3 признаков; высокий риск — наличие 4 признаков и более. При низком и промежуточном риске изначально показана монохимиотерапия, при высоком риске с самого начала рекомендуется полихимиотерапия (как следует из схемы 23, полихимиотерапию назначают также в IIIстадии болезни). В развернутой стадии проводится курсовая химиотерапия. Используют тот же препарат, но в больших дозах (ежедневно 2 — 3 приема) под гематологическим контролем: при снижении количества лейкоцитов и тромбоцитов дозу препарата уменьшают, а при содержании лейкоцитов 10 —20109/л и тромбоцитов 100-109/л препарат отменяют. Если ранее эффективные препараты не оказывают действия в течение 3 — 4 нед, то следует провести курс лечения другим цитостатиком. Так, если гид pea оказывается неэффективной, то назначают миелосан (бусульфан, милеран), миелобромол. • После курсовой химиотерапии проводится поддерживающая тера • Полихимиотерапия проводится курсами при высокой степени • В настоящее время появились принципиально новые методы лече На практике используют рекомбинантный а-ИФН— реаферон, или Интрон А, который вводится внутримышечно или подкожно. При лечении этим препаратом возможно появление «гриппоподобного» синдрома — по- вышение температуры, головная боль, ломота в мышцах, общее плохое самочувствие, однако прием парацетамола купирует эти явления. Реаферон, интрон А иногда комбинируют с цитостатическим препаратом — гидреа или цитозин-арабинозидом (цитозаром), что позволяет улучшить результаты лечения; 5-летняя выживаемость при лечении Ин-троном А 82 —89 мес (у 50 % больных), тогда как при лечении миелосаном этот показатель равен 44 — 48 мес. Весьма существенно, что при лечении а-ИФН может наступить не только гематологическая, но и цитогенетическая ремиссия, когда в клетках крови и костного мозга Ph-хромосома вообще не определяется, что позволяет говорить не столько о ремиссии, сколько о полном выздоровлении отХМЛ. • При значительном увеличении селезенки иногда осуществляют об • При гнойно-воспалительных осложнениях проводят антибиотикоте- Прогноз.Длительность жизни больных ХМЛ в среднем составляет 3 — 5 лет, у отдельных больных достигает 7 — 8 лет. Профилактика.Точных мер предупреждения ХМЛ не существует, в связи с чем можно говорить лишь о вторичной профилактике болезни, которая состоит в предупреждении обострений болезни (поддерживающая терапия, исключение инсоляции, простудных заболеваний и пр.). Эритремия является миелопролиферативным заболеванием, хроническим, доброкачественно текущим лейкозом, при котором наблюдается повышенное образование эритроцитов, а также нейтрофильных лейкоцитов и тромбоцитов. Источник опухолевого роста — клетка-предшественница миелопоэза. Заболеваемость эритремией составляет около 0,6 на 10 000 населения. Одинаково часто болеют как мужчины, так и женщины. Эритремия является болезнью лиц пожилого возраста: средний возраст заболевших 55 — 60 лет, однако заболевание возможно в любом возрасте. Этиология.Причины развития заболевания неизвестны. Патогенез.В основе заболевания лежит опухолевая пролиферация всех трех ростков кроветворения — красного, гранулоцитарного и мегака-риоцитарного, однако доминирует рост красного ростка. В связи с этим основным субстратом опухоли являются созревающие в избыточном количестве эритроциты. Появляются очаги кроветворения в селезенке и печени. Увеличенное количество эритроцитов и тромбоцитов в периферической крови снижает скорость кровотока, повышает вязкость и свертываемость крови, что обусловливает появление ряда клинических симптомов. Классификация.Учитывают стадию течения процесса, вовлечение в патологический процесс селезенки и последующую трансформацию эрит-ремии в другие заболевания системы крови. Стадия I — начальная: содержание гемоглобина на верхней границе нормы, небольшое увеличение массы циркулирующих эритроцитов, селезенка увеличена незначительно или без изменений. АД нормальное или слегка повышено, очаговая гиперплазия костного мозга в трепанате из подвздошной кости. Стадия II — развернутая: фаза А — без миелоидной метаплазии селезенки (простой вариант плеторы без спленомегалии). Тотальная трех-ростковая гиперплазия костного мозга. Отсутствие экстрамедуллярного гемопоэза; фаза Б — с миелоидной метаплазией селезенки. Большой мие-лопролиферативный синдром: панцитоз в периферической крови, в костном мозге имеется панмиелоз с очаговым миелофиброзом или без него, миелоидная метаплазия селезенки с фиброзом или без него. Стадия III — терминальная: перерождение доброкачественной опухоли в злокачественную (миелофиброз с анемизацией, хронический мие-лолейкоз, острый лейкоз). Миелофиброз развивается практически у всех, болеющих более 10—15 лет; он отражает естественную эволюцию болезни. Признаком миелофиброза является цитопения (анемия, тромбоцитопения, реже — лейкопения). Развитие хронического миелолейкоза проявляется нарастанием лейкоцитоза, увеличением (или появлением) в периферической крови клеток гранулоцитарного ряда (миелоцитов, промиелоцитов), а также обнаружением в клетках крови и костного мозга Ph-хромосомы. Острый лейкоз развивается обычно у больных, леченных цитостати-ками и радиоактивным фосфором. Анемия у больных эритремией может быть связана с частыми кровопусканиями, возможно повышенное депонирование эритроцитов, а также гемолиз эритроцитов. Клиническая картина.Эритремия проявляется двумя большими синдромами. Плеторический синдром обусловлен увеличенным содержанием эритроцитов, а также лейкоцитов и тромбоцитов (плетора — полнокровие). Этот синдром складывается из: 1) субъективных синдромов, 2) нарушений сердечно-сосудистой системы, 3) сдвигов в лабораторных показателях. 1. К субъективным симптомам плеторического синдрома относятся: 2. Нарушения сердечно-сосудистой системы проявляются в измене 3. Сдвиги в лабораторных показателях определяются главным обра гемоглобина и эритроцитов, повышение показателя гематокрита и вязкости крови, умеренный лейкоцитоз со сдвигом лейкоцитарной формулы влево, тромбоцитоз, резкое замедление СОЭ. Миелопролиферативный синдром обусловлен гиперплазией всех трех ростков кроветворения в костном мозге и экстрамедуллярно. Он включает: 1) субъективные симптомы, 2) спленомегалию и(или) гепатомегалию, 3) изменения лабораторных показателей. 1. К субъективным симптомам относятся слабость, потливость, по 2. Спленомегалия (увеличение селезенки) объясняется не только мие- 3. Среди изменений лабораторных показателей наибольшее диагнос Различная выраженность синдромов на разных стадиях болезни обусловливает чрезвычайную вариабельность клинической картины. Можно наблюдать больных с несомненной эритремией, почти не предъявляющих жалоб и полностью трудоспособных, и больных с тяжелым поражением внутренних органов, нуждающихся в проведении терапии и утративших трудоспособность. На I этапе диагностического поиска в начальной стадии заболевания больные могут не предъявлять никаких жалоб. По мере прогрессиро-вания болезни жалобы связаны с наличием и выраженностью плеторы и миелопролиферативного процесса. Наиболее часты жалобы «плеторического» характера, обусловленные повышенным кровенаполнением сосудов и функциональными нейрососудистыми расстройствами (головные боли, эритромелалгия, нарушение зрения и пр.). Вся эта симптоматика может быть связана и с другими заболеваниями, что необходимо выяснить при дальнейшем обследовании больного. Жалобы, обусловленные наличием миелопролиферативного синдрома (потливость, тяжесть в левом подреберье, боли в костях, повышение температуры тела) также неспецифичны для эритремии. Достаточно характерен кожный зуд, который появляется после приема водных процедур. Этот симптом наблюдается у 55 % больных в развернутой стадии и объясняется гиперпродукцией базофилов и гистаминемией. Аналогична природа крапивницы, наблюдающейся у 5 —7 % больных. Перечисленные симптомы имеют значение для определения стадии эритремии: их наличие обычно указывает на переход во ПБ стадию или в терминальную стадию с развитием миелофиброза как наиболее частого исхода эритремии. В анамнезе больных могут быть мозговые инсульты, инфаркты миокарда — все это свидетельствует об осложнениях заболевания. Иногда болезнь дебютирует именно этими осложнениями, а истинная причина их развития — эритремия — выявляется при обследовании больного по поводу инсульта или инфаркта миокарда. Указания на проводимое ранее лечение радиоактивным фосфором, цитостатиками или кровопусканиями могут навести на мысль о наличии какого-либо опухолевого заболевания крови. Уменьшение симптоматики плеторического синдрома на фоне лечения указанными средствами позволяет предположить эритремию. На II этапе диагностического поиска можно выявить отчетливые симптомы лишь во II (развернутой) стадии болезни. Обнаруживают в основном признаки плеторического синдрома: эритроцианоз, инъецированные сосуды конъюнктивы («кроличьи глаза»), отчетливая цветовая граница в месте перехода твердого неба в мягкое. Можно выявить симптомы эритромелалгии: отек кончиков пальцев, стоп, нижней трети голени, сопровождающийся локальной гиперемией и резким жжением. При исследовании сердечно-сосудистой системы диагностируют АГ и увеличение левого желудочка, в развернутой стадии болезни — «пестрые ноги» — изменение кожных покровов голеней (преимущественно дисталь-ной их части) в виде различной интенсивности участков пигментации, обусловленных нарушением венозного кровообращения. При пальпации живота можно обнаружить увеличение селезенки, что является одним из характерных признаков болезни. Увеличение селезенки может быть обусловлено: 1) повышенным депонированием элементов крови; 2) «рабочей» гипертрофией вследствие увеличения ее секвестрирующей функции; 3) экстрамедуллярным кроветворением (миелоидная метаплазия с преобладанием эритропоэза). Эти причины часто сочетаются. Обнаруживаемое иногда увеличение печени также обусловлено аналогичными причинами, а также развитием фиброза и хронического холецистита (неспецифический реактивный гепатит). Следует иметь в виду, что гепатомегалия может наблюдаться при злокачественной опухоли печени с развитием вторичного эритроцитоза. Осложнения эритремии в виде тромбозов сосудов головного мозга выражаются рядом очаговых симптомов, выявляемых при исследовании ЦНС. Однако и на II этапе поставить окончательно диагноз эритремии нельзя, так как многие ее симптомы могут встречаться при симптоматических эритроцитозах. Кроме того, отдельно взятые симптомы, такие как АГ, спленомегалия и гепатомегалия, характерны для самых разнообразных заболеваний. В связи с этим III этап диагностического поиска приобретает решающее значение, так как позволяет: а) поставить окончательный диагноз; б) уточнить стадию эритремии; в) выявить осложнения; г) осуществить контроль за лечением. Анализ периферической крови обнаруживает эритроцитоз, увеличение содержания гемоглобина и показателя гематокрита, что, однако, встречается и при симптоматических эритроцитозах. Для диагноза значение имеет сочетание повышения уровня гемоглобина с эритроцитозом, лейкоцитозом и тромбоцитозом. При исследовании лейкоцитарной формулы обнаруживают нейтрофилез и иногда незрелые гранулоциты. Если изменения в периферической крови незначительны или данные неубедительны (например, эритроцитоз не сочетается с тромбоцитозом), то необходимо провести исследование костного мозга (трепанобиопсия). Наличие в трепанате тотальной трехростковой гиперплазии костного мозга с преобладанием эритропоэза, замещение жировой ткани красным костным мозгом дают возможность поставить окончательный диагноз. Расширение «плацдарма» кроветворения выявляется также с помощью радионуклидного ска- нирования костей с 32Р. Гистохимическое исследование выявляет повышенную активность щелочной фосфатазы нейтрофилов. Осложнения. Течение эритремии осложняют: 1) сосудистые тромбозы (мозговых, коронарных, периферических артерий); 2) геморрагический синдром: кровотечения после малых оперативных вмешательств (экстракция зуба), из сосудов пищеварительного тракта, геморроидальных узлов, что обусловлено плохой ретракцией кровяного сгустка вследствие изменения функциональных свойств тромбоцитов; 3) эндогенная урикемия и урикурия (вследствие повышенной гибели клеток на ядерных предстадиях их созревания), что проявляется симптомами мочекаменной болезни и подагрического артрита. Исходами болезни являются ситуации, указанные в III стадии течения болезни (миелофиброз, хронический миелолейкоз, острый лейкоз, анемия). Диагностика. Эритремию можно заподозрить у лиц с наличием стойкого эритроцитоза в сочетании с нейтрофильным лейкоцитозом, тромбоци-тозом при отсутствии заболеваний (или состояний), которые могли бы вызвать эритроцитоз. Диагностическими критериями эритремии (в развернутой стадии) являются: • увеличение числа эритроцитов в крови (более 61О12/л у мужчин и • увеличение содержания НЬ (более 177 г/л для мужчин и более • увеличение показателей гематокрита (52 % для мужчин и 48 % для увеличение массы циркулирующих эритроцитов; лейкоцитоз более 12109/л (при отсутствии явных причин для появления лейкоцитоза); тромбоцитоз более 400-109/л; увеличение абсолютного числа базофилов в крови; увеличение селезенки; нормальное насыщение артериальной крови кислородом (более 92 %); трехростковая пролиферация в костном мозге (по данным трепано-биопсии) с вытеснением из него жира; • нормальное содержание эритропоэтина в крови. Затруднения в постановке диагноза обусловлены развитием так называемого симптоматического эритроцитоза при целом ряде заболеваний. Выделяют абсолютные и относительные симптоматические эритроцитозы. При абсолютных эритроцитозах отмечаются увеличение массы циркулирующих эритроцитов и повышенный эритропоэз. Для относительных эрит-роцитозов характерны уменьшение объема циркулирующей плазмы и постоянство массы циркулирующих эритроцитов. Причины развития симптоматических эритроцитозов: 1) генерализованная тканевая гипоксия (легочная патология, заболевания сердца, гемоглобинопатии, ожирение и т.д.); 2) паранеопластические реакции (опухоли почек, опухоли коркового и мозгового вещества слоя надпочечников, гипофиза, яичников, сосудистые опухоли, опухоли других органов); 3) ишемия почек (стеноз почечной артерии, гидронефроз, поликистоз и другие аномалии почек); 4) неустановленные причины (заболевание ЦНС, портальная гипертензия). Относительные эритроцитозы наблюдаются при эксикозах (обезвоживание вследствие поноса, рвоты, повышенной потливости и др.). Дифференциальная диагностика основывается на учете всей клинической картины. В сложных случаях необходимо исследовать содержание эритропоэти-на в крови; при эритремии оно повышается. Формулировка развернутого клинического диагнозавключает сведения: 1) о стадии заболевания; 2) о наличии осложнений; 3) о фазе процесса (обострение или ремиссия); 4) о наличии выраженных синдромов (портальная гипертензия, АГ и пр.). Лечение.Весь комплекс лечебных мероприятий при эритремии представлен в табл. 15. Таблица 15. Комплекс лечебных мероприятий при эритремии
Основные направления терапии Лечебные средства и мероприятия Ликвидация плеторы Кровопускания, дезагреганты Борьба с миелоидной пролиферацией Цитостатическая терапия Лечение исходов болезни:
миелофиброза Цитостатическая терапия, гемотрансфузии,
спленэктомия острого лейкоза Полихимиотерапия хронического миелолейкоза Цитостатическая терапия Лечение осложнений:
сосудистых тромбозов Антикоагулянты, дезагреганты портальной гипертензии Салуретики, антагонисты альдостерона В развернутой стадии болезни при наличии плеторического синдрома, но без лейко- и тромбоцитоза применяют кровопускания как самостоятельный метод терапии. Извлекают по 400 — 500 мл крови за один раз через день (в условиях стационара) или через 2 дня (в условиях поликлиники). Для профилактики тромбозов (развивающихся в результате кровопускания) назначают ацетилсалициловую кислоту в дозе 0,5— 1 г/сут накануне и в день кровопускания, а также в течение 1 — 2 нед после окончания кровопусканий. Кроме ацетилсалициловой кислоты, назначают и другие дезагреганты — дипиридамол (курантил), тиклоридин (тиклид), трентал одновременно с аспирином. Перед кровопусканием целесообразно ввести внутривенно 400 мл реополиглюкина, а также 5000 ЕД гепарина (через иглу Дюфо). При плохой переносимости кровопусканий, наблюдаемой у лиц с выраженным атеросклерозом мозговых сосудов, ограничиваются эксфузией 300 мл (2 раза в неделю). При кровопусканиях необходимо снизить гемоглобин до 150 г/л, а показатель гематокрита до 42 — 47 %. Если кровопускания недостаточно эффективны, а также при формах болезни, протекающих с панцитозом и спленомегалией, назначают цитостатическую терапию. Возраст больных более 55 лет рас- ширяет показания к применению цитостатиков. Косвенными показаниями к цитостатической терапии являются и другие признаки миелопролиферативного синдрома (зуд), а также тяжесть заболевания, висцеральные сосудистые осложнения (инсульт, инфаркт миокарда), истощение. Противопоказаниями к цитостатической терапии являются молодой возраст больных, рефрактерность к лечению на предыдущих этапах, а также чрезмерно активная в прошлом цитостатическая терапия из-за опасения перехода заболевания в фазу анемии. Эффект цитостатической терапии следует оценивать через 3 мес после окончания лечения; это объясняется тем, что продуцированные до лечения эритроциты живут в среднем около 2 — 3 мес. Снижение количества лейкоцитов и тромбоцитов наступает значительно раньше, соответственно срокам их жизни. Критерием эффективности цитостатической терапии является достижение гематологической ремиссии (полной, когда все показатели крови нормализуются, или частичной, при которой остается несколько повышенным количество эритроцитов, лейкоцитов и/или тромбоцитов). Из цитостатических препаратов на первом этапе обычно назначают гидроксимочевину (гидреа) под контролем периферической крови (1 раз в 2 нед). Препарат обладает незначительным мутагенным эффектом: под его воздействием не отмечено увеличения частоты развития лейкозов. Лечение гидроксимочевиной следует проводить постоянно, так как с точки зрения эффективности этот препарат уступает другим препаратам (алки-лирующим агентам — миелосан, миелобромол и др.) по силе воздействия на эритроидный росток; кроме того, при лечении гидроксимочевиной продолжают кровопускания. Однако если этот препарат не дает достаточного эффекта, то используют группу алкилирующих препаратов. Из всего арсенала препаратов этой группы предпочтение отдается нитрозомочевине (препарат назначается курсами до 1 мес и вводится внутривенно), так как при длительном использовании ранее применявшихся алкилирующих препаратов отмечался несколько более частый исход эритремии в ОЛ. Тем не менее миелосан и миелобромол остаются в арсенале врача при лечении эритремии. Весьма существенно, что рецидивы заболевания следует лечить тем же цитостатиком, который вызвал ремиссию. Назначение нового препарата должно происходить при неэффективности предшествующего лечения или появлении нового качества болезни (например, развитие ХМЛ). • На исходы эритремии (миелофиброз, острый лейкоз, хронический • Симптоматическую терапию при приступах эритромелалгии прово • При осложнениях эритремии тромбозом сосудов применяют антико- • Больных эритремией ставят на диспансерный учет с частотой обращения к врачу и назначением анализов периферической крови 1 раз в 3 мес. Прогноз.При неосложненном течении эритремии продолжительность жизни может достигать 15 — 20 лет (в дальнейшем возникают осложнения). Если же осложнения со стороны сердечно-сосудистой системы развиваются достаточно рано или же болезнь прогрессирует, продолжительность жизни сокращается. Во всяком случае при прочих равных условиях своевременно начатая терапия удлиняет продолжительность жизни, хотя это наблюдается не во всех случаях. Профилактика.Радикальных мер предупреждения болезни не существует, в связи с чем можно говорить лишь о вторичной профилактике, заключающейся в динамическом наблюдении за больными и проведении противорецидивной терапии.
|
|||||||||
|
|
Последнее изменение этой страницы: 2024-06-27; просмотров: 5; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.222.67.8 (0.037 с.) |

 Содержание
Содержание