Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Теория возмущения мёллера-плессетаСодержание книги
Поиск на нашем сайте
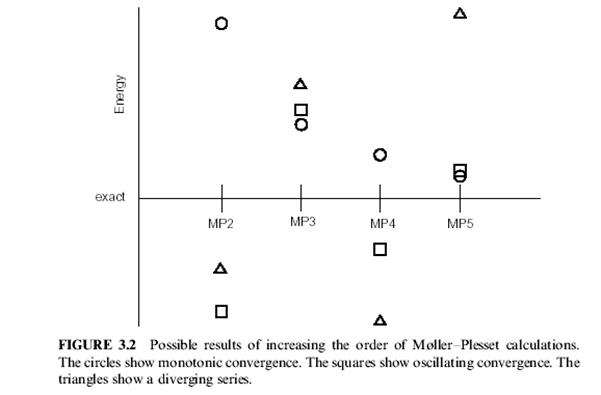
Корреляция может быть добавлена как пертурбация для волновой функции Хартри-Фока. Это называется теорией возмущения Мёллер-Плессета. В отображении волновой функции HF в формулировке теории возмущения, HF становится возмущением первого порядка. Таким образом, минимальное количество корреляции, которое будет добавлено, использует второй порядок - метод MP2. (Третий порядок - MP3, четвертый порядок - MP4). Точность вычисления MP4 почти эквивалентна точности CISD вычисления. MP5 и высшие порядки редко применяются из-за высокой вычислительной стоимости (N10 затрат времени или больше). Вычисления Мёллер-Плессета - не вариационны. Фактически, вычисления MP2, дают полные энергии ниже точных значений полной энергии. В зависимости от природы химической системы, можно использовать последовательно Высшие порядки из теории возмущения. Для некоторых систем, энергии, полученные последовательно приближаются к полной энергии в ряду от MP2 до MP3,и к MP4, и так далее, как показано на рисунке 3.2. Для других систем, MP2 будет иметь энергию ниже чем точная энергия, MP3 будет выше, MP4 будет ниже, и так далее, с каждым наличием ошибки, которая является одинаковой по величине, но противоположной по знаку. Если предположение о малой возмущения не имеет силу, MPn энергии могут отклоняться, как показано на рисунке 3.2. Одно из преимуществ Мёллер-Плессета - обширный размер используемых систем. Разработан также локальный MP2 метод (LMP2). LMP2 вычисления требуют меньшего количества времени CPU, чем вычисления MP2. LMP2 - также менее склонен к ошибке наложения (суперпозиции) базисных наборов. Цена этих усовершенствований равна приблизительно 98 % MP2 от исправления энергии полученной из LMP2. КОНФИГУРАЦИОННОЕ ВЗАИМОДЕЙСТВИЕ Конфигурационное взаимодействие волновой функции - мультидетерминант волновой функции. Он может быть создан, начиная с волновой функция HF и создавая новые детерминанты при промотировании электронов с занятых на незанятые орбитали. Вычисления с помощью конфигурационного взаимодействия могут быть очень точны, но стоимость времени CPU очень высока (N8 увеличение времени или больше).Вычисления конфигурационного взаимодействия классифицируются числом возбуждений, которое обыкновенно может делать каждый детерминант. Если был перемещен только один электрон для каждого детерминанта, то это называется вычислением - отдельно возбужденным конфигурационным взаимодействием (CIS). Вычисления CIS дают аппроксимацию возбужденного состояния молекулы, но не изменяют энергию основного состояния. Вычисление отдельного - и двойного возбуждения (CISD) дают энергию основного состояние, которая была исправлена корреляцией. Вычисления тройного возбуждения (CISDT) и учетверенного возбуждения (CISDTQ) могут быть сделаны только тогда, когда требуются результаты очень высокой точности. Вычисление конфигурационного взаимодействия со всеми возможными возбуждениями называется полным CI. Полное CI вычисление, использующее бесконечно большой базисный набор даст точный квантовомеханический результат. Однако, полные CI вычисления делаются очень редко из-за огромного количества требуемой компьютерной мощи. Результаты CI могут немного изменяться при переходе от одной программы к другой, для молекул с открытой оболочкой. Это происходит из-за используемого стандартного состояния HF. Некоторые программы, типа Gaussian, используют UHF стандартное состояние. Другие программы, типа MOLPRO и MOLCAS, используют ROHF стандартное состояние. Различие в результатах обычно довольно малое и становится еще меньше с использованием вычисления высшего порядка. При использовании полного CI нет никакого различия.
|
||||
|
|
Последнее изменение этой страницы: 2017-01-19; просмотров: 387; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.101.75 (0.009 с.) |