Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Методы линейного масштабирования
Одни из последних усовершенствований в DFT - появление алгоритмов линейного масштабирования. Эти алгоритмы заменяют Кулоновские взаимодействия для отдаленных областей молекулы на взаимодействия мультиполей. Эти результаты позволяют проводить быстрые расчеты для достаточно больших молекул. Наиболее общие линейные методы масштабирования - метод быстрых мультиполей (FMM) и непрерывный быстрый мультипольный метод (CFMM). DFT обычно быстрее, чем метод Хартри-Фока для систем больше чем с 10-15 неводородными атомами, в зависимости от точности чисел и базисного набора. Алгоритмы линейного масштабирования станут выгодными, когда число тяжелых атомов превышает 30 или больше, в зависимости от общей формы молекулы. Методы линейного масштабирования DFT вероятно самые быстрые ab initio методы для больших молекул. Однако в литературе имеется много заблуждений на этот случай. Достоверная литература с графиками, указывает, что линейные методы масштабирования требуют величины времени CPU на порядок, чем обычные алгоритмы для некоторых испытательных систем, типа n - плоскостей графита или алканов. Однако, вычисления с коммерческим программным обеспечением часто указывают ускорение работы только на несколько процентов или возможно даже слегка более медленное вычисление. Есть множество причин для этих несообразностей. Первый фактор- большинство пакетов программ, разработанных для эффективного действия, использует сокращения в точности по сравнению с ab initio вычислениями. Это означает, что интегралы, включающие отдаленные атомы не включены в вычисление, если они имеют незначительное воздействие на вычисление конечной энергии, что обычно меньше чем 0.00001 Хартри или сотую энергии взаимодействия Ван-дер-Ваальса. Многие из графиков, показывая выполнение линейного масштабирования DFT, сравнивают его с алгоритмом, который не использует составные сокращения точности. Случается, что вычисление выполняется быстрее из-за сокращения точности метода линейного масштабирования. Второе соображение - геометрия молекулы. Методы оценки мультиполей имеют силу только для описания взаимодействий между отдаленными областями молекулы. Линейные системы могут быть смоделированы наиболее эффективно, но шаровидные или плоские системы менее эффективно. В наших испытательных вычислениях на н-алкане C40H82, вычисление энергии в свернутой конформации потребовало в четыре раза больше времени CPU, чем вычисление в линейной конформации.
По сути, эти методы линейного масштабирования могут использовать меньшее количество времени CPU, чем обычные методы, но ускорение не настолько большое, как описано в литературе. Мы проводили испытательные вычисления на н-алкане C40 в различных конформациях. Эти вычисления показали, что методы линейного масштабирования требовали 60-80 % от количества времени CPU, требуемого для обычного вычисления. Можно получить ускорение выполнения больше чем это, вручную регулируя используемый алгоритм, но исследователи дают критическое предостережение о выполнении этого, потому что это может затронуть точность результатов. ПРАКТИЧЕСКИЕ СООБРАЖЕНИЯ Как упомянуто выше, DFT вычисления должны использовать базисный набор. Возникает вопрос о том, какие DFT оптимизированные или типичные HF оптимизированные базисные наборы должны использоваться. Использование DFT - оптимизированных базисных наборов не показало никакого преимущества перед обычными базисными наборами. Большинство DFT вычислений сегодня сделается с HF - оптимизированными GTO базисными наборами. Точность результатов имеет тенденцию значительно ухудшаться с использованием очень малого базисного набора. Из соображений точности, самый маленький используемый базисный набор - обычно 6-31G* или эквивалентный ему. Интересно, имеется только малое увеличение в точности, полученной, используя очень большие базисные наборы. Это, вероятно, происходит вследствие того, что Функционал плотности ограничивает точность больше, чем ограничения базисных наборов. DFT вычисления используют числовые интегралы базисных GTO. Можно ожидать, что STO базисные наборы или числовые базисные наборы (например, кубические плавные кривые) были бы более точны из-за правильного представления ядерного остова и показательного затухания в длинных расстояниях. Факт, что так много DFT используют базисные наборов GTO– это не отражение точности или преимуществ времени вычисления. Это произошло потому, что большое количество программ, было написано для GTO вычислений HF. Программы HF могут быть легко превращены в DFT программы, так что имеется много общего между этими программами. Есть программы, которые используют и кубические искривленные базисные наборы (например, dMol и программы Spartan) и STO базисные наборы (например, ADF).
Точность результатов из DFT вычислений будет находиться в зависимости от выбора базисного набора и функционала плотности. Выбор функционала плотности сделать трудно, поэтому идет создание новых функционалов. Во время публикации этой книги, B3LYP функциональный гибрид (также называемый Becke3LYP) был наиболее широко использован для молекулярных вычислений из-за точности результатов B3LYP, полученных для большого разнообразия соединений, особенно органических молекул. Однако, не будет удивительно если это господство функционала изменится в течение нескольких лет. Таблица 5.1 содержит список множества обычно используемых функционалов. Из-за новизны DFT, его выполнение еще не известно полностью и продолжает изменяться с развитием новых функционалов. Библиография в конце этой главы включает рекомендации для изучения и сравнения точности результатов. В настоящее время, результаты DFT очень хороши для органических молекул, особенно с закрытыми оболочками. Результаты не были настолько ободрительны для тяжелых элементов, высоко заряженных систем или систем очень чувствительных к электронной корреляции. Также, функционалы, внесенные в список в Таблице 5.1 и не решают проблемы возникающие из- за дисперсионных сил. РЕКОМЕНДАЦИИ Учитывая факт, что DFT является более новым, чем другие ab initio методы, вероятно, что в не слишком отдаленном будущем появятся новые методики. Функционалы плотности часто использовались из-за оптимальной точности с улучшением времени CPU. Во время публикации этой книги, для вычислений многих органических молекул широко использовался метод B3LYP с базисным набором 6-31G* или большим. К сожалению, нет никакого конкретного единого способа улучшить DFT вычисления, что делает их непригодными для работы очень высокой точности. Исследователи рекомендуют осуществить литературные поиски и выполнять предварительные испытательные вычисления перед использованием этих методов.
Молекулярная механика Наиболее серьезное ограничение ab initio методов - ограниченный размер молекулы, которую можно смоделировать даже на самых больших компьютерах. Полуэмпирические вычисления могут использоваться для больших органических молекул, но также слишком долгие для больших биомолекулярных систем. Если молекула настолько большая, что полуэмпирическая обработка не может использоваться эффективно, возможно моделировать её поведение, исключающее квантовую механику полностью, и используя молекулярную механику. ОСНОВЫ ТЕОРИИ Выражение энергии в молекулярной механике состоит из простого алгебраического уравнения для энергии вещества. Оно не использует волновую функцию или полную электронную плотность. Константы в этом уравнении получены или из данных спектроскопии или вычислений ab initio. Система уравнений с их связанными константами называется силовым полем. Фундаментальное предположение о методе молекулярной механики - переносимость параметров. Другими словами, проблема энергии, связана со специфическим молекулярным движением, скажем, растяжением одинарной связи углерод-углерод, и будет та же самая от одной молекулы до следующей. Это дает очень простое вычисление, которое может применяться к очень большим молекулярным системам. Работа этой методики зависит от четырех коэффициентов:
1. Функциональная формы выражения энергии 2. Данные, которые использовали в параметризированных константах 3. Методика имеет обыкновение оптимизировать константы из этих данных 4. Возможность пользователя, изменить методику на пути, в зависимости от его желания Для переносимости параметров, чтобы хорошо описывать молекулы, силовые поля используют типы атома. Это означает, что sp3 углерод будет описан другими параметрами, чем sp2 углерод и так далее. Обычно, атомы в ароматических кольцах будут обработаны по-другому в отличие от sp2 атомов. Некоторые силовые поля имеют даже параметризированные атомы для определенных функциональных групп. Например, карбонил. Карбонильный кислород в карбоновой кислоте может быть описан другими параметрами, чем кислород карбонила в кетоне. Выражение энергии состоит из суммы простых классических уравнений. Эти уравнения описывают различные аспекты молекулы, типа длины связи, изгиба связи, скручиваний, электростатических взаимодействий, сил Ван-дер-Ваальса и водородной связи. Силовые поля отличаются по числу условий для выражения энергии, сложности этих условий, и пути, по которым были получены константы. Так как электроны явно не включены, электронные процессы не могут быть смоделированы. Условия для выражения энергии, которые описывают отдельный аспект формы макромолекул, типа длины связи, изгиба углов, инверсии колец или торсионного движения, называются валентными условиями. Все силовые поля имеют, по крайней мере, одно валентное условие, а чаще всего имеют три или больше. Условия для выражения энергии, которые описывают, как одно движение молекулы затрагивает другое, называются взаимными условиями. Термин Взаимный, обычно используемый - термин изгиба растяжения, который описывает, равновесную длину связи, имеет тенденцию изменяться, как только углы связи будут изменены. Некоторые силовые поля не имеют никаких взаимных условий и могут компенсировать их наличием сложных электростатических функций. Силовое поле MM4 - в экстремальном значении проходит с девятью различными типами взаимных условий.
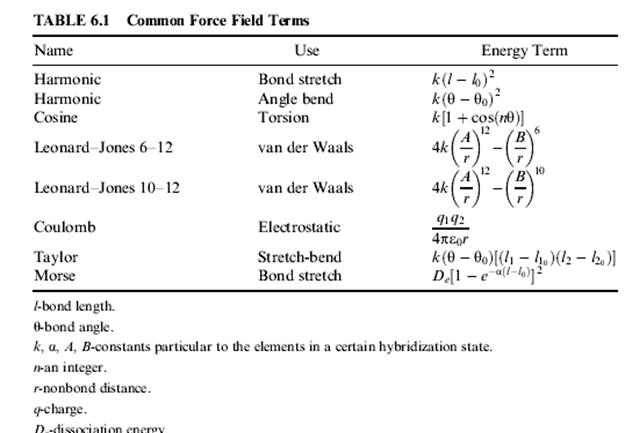
Силовые поля могут включать или не включать электростатические условия. Электростатический - наиболее часто используемый термин из закона Кулона для энергии притяжения или отталкивания между заряженными центрами. Обычно этот заряд получается из неорбитальных алгоритмов, разработанных для использования с молекулярной механикой, и как предполагается, является частичным зарядом на ядре. Моделирование молекул с чистым зарядом может быть лучше всего описано, используя типы атомов, параметризированных для описания заряженных центров. Иногда чтобы моделировать эффекты сольватации, включается диэлектрическая проницаемость. Эти методы вычисления зарядов описаны далее в Главе 12. Длина связи наиболее часто описывается уравнением гармонического осциллятора. Но иногда она описывается потенциалом Морзе. В редких случаях, длина связи может быть описана в уравнении потенциалом четвертой степени или потенциалом Леннарда-Джонса. Кубические уравнения использовались для описания длины связи, но они страдают тем, что плохо отражают отталкивания. Изгиб связи наиболее часто описывается гармоническим уравнением. Вращение связи обычно описывается выражением косинуса (Figure 6.1). Таблица 6.1 дает математические зависимости энергии, часто используемые в популярных силовых полях. Константы могут измениться от одного силового поля до другого, согласно выбору проектировщика системы единиц, нулевой энергии и процедуры приспособления. Все константы в этих уравнениях были получены из экспериментальных данных или ab initio вычислений. Метод молекулярной механики может быть, параметризован и для определенного класса молекул, типа белков или нуклеотидов. Такое силовое поле, будет уместным при описании других белков или нуклеотидов. Другие силовые поля - параметризуют, чтобы дать приемлемое описание различным классам органических соединений. Несколько силовых полей были даже параметризованы для всех элементов. Некоторые силовые поля упрощают сложные вычисления, опуская большинство водородных атомов. Параметры, описывающие каждый атом основной цепи, тогда будут изменены, чтобы описать поведение атомов с присоединенными (концевыми) водородами. Таким образом, вычисление вероятнее использует группу CH2, чем sp3 углерод, связанный с двумя водородами. Такие поля называются объединенными силовыми полями атома. Это вычисление наиболее часто используется, чтобы описать очень большие биомолекулы. Оно обычно не применяется, если доступные компьютерные аппаратные средства ЭВМ способны к использованию более точных явных водородных силовых полей. Некоторые силовые поля имеют типы атома для использования и с Неявными и Видимыми Водородами.
Также важен путь, по которым получены параметры силового поля из этих оригинальных данных. Длина связи и изгиб - относительно крутые движения. Таким образом, они могут часто описываться очень хорошо, используя равновесные значения, полученные из результатов дифракции Рентгена и силовых констант из колебательной спектроскопии. С другой стороны, торсионное поведение чувствительно, и к торсионному поведению конкретной связи и несвязанных взаимодействий между отдаленными частями молекулы и даже окружающими молекулами. Выбор процедуры применения становится важным, потому что он определяет, сколько части из энергии вложено от каждого процесса. Силовое поле может также быть, параметризовано лучше, предсказывая колебательное движение или силы межмолекулярного взаимодействия. Энергии, вычисленные молекулярной механикой – это обычно конформационные энергии. Вычисленная энергия, как предполагается, является энергией, которая надежно предскажет различие в энергии от одной конформации к другой. Эффект растяжения длин связи или углов также включен в эту энергию. Но это - не те же самые полные энергии, полученные из программ ab initio или теплоты образования из полуэмпирических программ. Фактическое значение конформационной энергии не обязательно имеет правильное физическое значение и - не сопоставимо между различными силовыми полями. Методы Молекулярной механики могут быть изменены для вычисления теплот образования с включением базы данных или схемы вычисления, чтобы выдать энергии связей, которые могли бы быть добавлены к конформационной энергии и пересчету на нуль энергии. Методы Молекулярной механики обычно не применимы к структурам, очень далеким от равновесия, типа переходных структур. Вычисления, которые используют алгебраические выражения, чтобы описать путь реакции и переходную структуру - обычно полуклассические алгоритмы. Эти вычисления используют выражение энергии, приспособленное к ab initio поверхности потенциальной энергии для конкретной реакции, скорее, чем использование тех же самых параметров для каждой молекулы. Полуклассические вычисления обсуждены далее в Главе 19. СУЩЕСТВУЮЩИЕ СИЛОВЫЕ ПОЛЯ Большинство исследователей самостоятельно не делает параметризированные силовые поля, потому что много хороших силовых поля уже было создано. В редких случаях, исследователь может присоединять дополнительный атом, как описано в Главе 29.. Многие из их были осуществлены в большом пакете программ. Имеется тенденция иметь незначительные различия в результатах вычислений при переходе от одного пакета программ до другого. AMBER Assisted model building with energy refinement (AMBER) (Вспомогательная модель, строящая с обработкой энергии) - название и, силовое поле программы молекулярной механики. Оно было определенно параметризовано для белков и нуклеиновых кислот. AMBER использует только пять связывающих и несвязывающих условий наряду со сложной электростатической обработкой. Никакие взаимные условия не включены. Результаты очень хороши для белков и нуклеиновых кислот, но могут быть несколько ошибочны для других систем. CHARMM Chemistry at Harvard macromolecular mechanics (CHARMm) (Химия в Гарвардской макромолекулярной механике) - название и силовое поле программы, включающей это силовое поле. Академическая версия этой программы обозначена, CHARMM, а коммерческая версия называются CHARMm. Оно было первоначально разработано для белков и нуклеиновых кислот. Теперь оно применяется для большого разнообразия биомолекул, сольватации, кристаллов, анализа колебаний и изучений QM/MM. CHARMm использует пять валентных условий, одно из которых - электростатическое. CFF The consistent force field (CFF) (согласованное силовое поле)-был развито, чтобы выдать согласованную точность результатов для конформаций, колебательных спектров, деформаций и колебательной энтальпии белков. Есть несколько вариаций этого типа: Ure-Bradley версия (UBCFF), валентная версия (CVFF), и Lynghy CFF. Квантовомеханически параметризированное силовое поле (QMFF), было параметризовано из ab initio результатов. CFF93 - перемасштабирование QMFF, чтобы воспроизвести экспериментальные результаты. Эти силовые поля используют пять из шести валентных условий, одно из которых - электростатический, и четыре из шести взаимных условий. CHEAT Carbohydrate hydroxyls represented by external atoms (CHEAT) (Гидроксилы Углеводов, представленны внешними атомами) - силовое поле, конкретно разработанное для моделирования углеводов. DREIDING DREIDING - универсальное силовое поле органических или биоорганических молекул. Оно наиболее широко использовалось для больших биомолекулярных систем и использует пять валентных условий, одно из которых - электростатический. Использование DREIDING уменьшилось с введением улучшенных методов. ECEPP Empirical conformational energy program for peptides (ECEPP) (Эмпирическая программа для конформационной энергии пептидов) - название компьютерной программы и силового поля, осуществленного в этой программе. Это - одно из более ранних силовых полей для пептидов, которое мало использовалось из-за введения улучшенных методов. Оно использует три валентных условия, которые связаны с силами ван-дер-Ваальса, и электростатическими взаимодействиями. EFF Empirical force field (EFF) –(эмпирическое силовое поле) силовое поле, разработанное только для моделирования углеводородов. Оно использует три валентных условия, никаких электростатических и пять взаимных условий. GROMOS Gronigen molecular simulation (GROMOS) (Гренинген молекулярное моделирование) - название силового поля и программы, включающей это силовое поле. GROMOS силовое поле популярно для предсказания динамического движения молекул и блочных жидкостей. Оно также используется для моделирования биомолекул. Оно использует пять валентных условий, одно из которых - электростатическое. MM1, MM2, MM3, MM4 MM1, MM2, MM3, и MM4 - универсальные органические силовые поля. Было много вариантов оригинальных методов, особенно MM2. MM1 редко используется, так как более новые версии показывают значительные усовершенствования. MM3 метод - вероятно один из наиболее точных путей моделирования углеводородов. Во время публикации этой книги, MM4 метод был только опубликован, и нет полных сведений о его результатах. Однако, начальные результаты ободрительны. Это - некоторые из наиболее широко используемых силовых полей из-за точности представления органических молекул. MMX и MMЗ - вариации MM2. Эти силовые поля используют пять из шести валентных условий, одно из которых - электростатическое и одно из девяти взаимных условий. MMFF Merck molecular force field (MMFF) –(молекулярное силовое поле Мерка) один из совсем недавно опубликованных силовых полей в литературе. Это - универсальный метод, особенно популярный для органических молекул. MMFF94 был первоначально предназначен для моделирования в молекулярной динамике, но также нашел использования при оптимизации геометрии. Он использует пять валентных условий, одно из которых - электростатическое и одно взаимное. MOMEC MOMEC - силовое поле для описания координационных соединений переходных металлов. Оно было, первоначально параметризовано, чтобы использовать четыре валентных условия, но не электростатическое условие. Взаимодействия металл - лиганд состоят из растяжения связи. Сфера координации поддерживается несвязанные взаимодействиями между лигандами. MOMEC обычно работает приемлемо хорошо для октаэдрически координированных соединений. OPLS Optimized potentials for liquid simulation (OPLS) (Оптимизация потенциалов для моделирования жидкости) был разработан для моделирования жидкостей. Оно также имело большое использование при моделировании молекулярной динамикой биомолекул. OPLS использует пять валентных условий, одно из которых - электростатическое, но никаких взаимных условий. Tripos Tripos - силовое поле, созданное в Tripos Inc для включения в Alchemy и SYBYL программы. Оно иногда называется SYBYL силовым полем. Tripos разработан для моделирования органических и биоорганических молекул. Оно также часто используется для CoMFA анализа, 3-D QSAR методики. Tripos использует пять валентных условий, одно из которых - электростатическое. UFF UFF - universal force field. Множество универсальных силовых полей, означают, что они включают все элементы,. Это - наиболее многообещающее полное силовое поле периодической таблицы, доступное в настоящее время. UFF наиболее широко используется для систем, содержащих неорганические элементы. Оно было разработано, чтобы использовать четыре валентных условия, но не электростатическое. Программное обеспечение рекомендует использовать заряды, полученные с Q-equilibrate методом. Наши независимые изучения нашли, что точность результатов, может быть значительно лучше без этого. YETI YETI - силовое поле, разработанное для точного представления несвязанных взаимодействий. Оно наиболее часто используется для моделирования взаимодействий между биомолекулами субстрата и малыми молекулами. Оно не предназначено для оптимизации молекулярной геометрии, так что исследователи часто оптимизируют молекулярную геометрию с другим силовым полем, типа AMBER, а затем используют YETI, чтобы моделировать процесс взаимодействия. Недавние присоединения к YETI - поддержка для металлов и эффектов растворителя. ПРАКТИЧЕСКИЕ СООБРАЖЕНИЯ Силовые поля для описания неорганических элементов еще не увидели такого развития как силовые поля органических молекул. Множество органических методов было расширено до полной применимости всей периодической таблицы, но результаты были не очень захватывающими. Лучшее доступное силовое поле для неорганики - вероятно UFF силовое поле. При изучении неорганики в прошлом не использовали существующие ранее силовые поля, типа UFF и часто делали параметризацию нового атома, чтобы описать поведение неорганического элемента в органическом силовом поле. Моделирование Неорганических веществ более подробно обсуждено в Главах 37 и 41. Вычисления Молекулярной механикой обманчиво просты. Много пакетов программ теперь делают молекулярную механику столь же легкой, как и определение молекулярного строения. Трудность находится в осознании, каким результатам можно доверять. Наиболее надежные результаты те, которые показывают различия энергии между конформерами. Другое популярное использование - исследование межмолекулярного взаимодействия. Различие в энергии связи между двумя участками или двумя ориентациями обычно показывается довольно надежно. Абсолютная энергия связи (разнесение молекул к бесконечности) будет предсказано не так надежно. Другие вычислительные свойства кроме энергии и геометрии будут обсуждены в главе 13. РЕКОМЕНДАЦИИ Преимущество молекулярной механики - позволяет моделирование огромных молекул, типа белков и сегментов ДНК. Поэтому она первичный инструмент вычислений для биохимиков. Она также хорошо моделирует силы межмолекулярного взаимодействия. Неудобство молекулярной механики там – где много химических свойств, которые даже не определены в пределах метода, типа возбужденных электронных состояний. Так как условия образования химической связи явно включены в силовое поле, оно не возможно без некоторых математических манипуляций, чтобы исследовать реакции, в которых связи будут сформированы или нарушены. Чтобы работать с чрезвычайно большими и сложными системами, пакеты программ молекулярной механики часто имеют мощные и легкие в использовании графические описатели. Из-за этого, механика иногда используется, так как это легкий, но не обязательно хороший, способ описать систему. Из-за чувствительности к параметризации, лучшая методика - выбора силового поля - предварительно поискать подобные расчеты в литературе и сверить полученные результаты с экспериментальными результатами. Публикации, внесенные в библиографический список в конце этой главы и в Главе 16 дают некоторые исходные превосходные точки для обнаружения и сравнения точности. Обобщение результатов для изучений, сравнивающих точности силовых полей следующие: 1. MM2, MM3, и Merck (MMFF) - силовые поля работают лучше всего для органических молекул. 2. AMBER и CHARMm - силовые поля дают лучшие результаты для изучения нуклеиновых кислот и белков. 3. Изучение молекулярной механикой неорганических молекул требуют осторожной настройки параметров силового поля. 4. UFF - наиболее надежное силовое поле, которое можно использовать без модификации для неорганических систем. 5. Изучение Молекулярной динамикой лучше делать с конкретным силовым полем, разработанным для этой цели. 6. Кольца сахаров ставят специфические проблемы в универсальных силовых полях и должны быть смоделированы, используя силовое поле, разработанное для углеводов.
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-01-19; просмотров: 993; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.21.248.47 (0.055 с.) |
 Силы межмолекулярного взаимодействия, типа взаимодействий Ван-дер-Ваальса и водородной связи, часто описываются уравнениями Леннарда-Джонса. Некоторые силовые поля также используют объединенное уравнение для изгиба и растяжения. Выбор функциональной формы уравнения особенно важен для вычисления энергий молекул, искаженных от геометрии равновесия, как показано различием между гармоническим потенциалом и потенциалом Morse, показанным на рисунке 6.2.
Силы межмолекулярного взаимодействия, типа взаимодействий Ван-дер-Ваальса и водородной связи, часто описываются уравнениями Леннарда-Джонса. Некоторые силовые поля также используют объединенное уравнение для изгиба и растяжения. Выбор функциональной формы уравнения особенно важен для вычисления энергий молекул, искаженных от геометрии равновесия, как показано различием между гармоническим потенциалом и потенциалом Morse, показанным на рисунке 6.2.