
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Релаксаційні процеси в наповнених полімерахСодержание книги
Поиск на нашем сайте
Перехід будь-якої системи з нерівноважного стану в рівноважний називається релаксацією. Якщо систему характеризують за зміною напруження, то говорять про релаксацію напруження (деформації, об’єму й т.д.). Якщо напруження в зразку − σ, то швидкість релаксації напруження − dσ/dt, тобто швидкість переходу до ненапруженого стану пропорційна напруженню:
де
Нехай t = τ, тоді
Чим гнучкіші макромолекули полімеру, тим коротша довжина кінетичного сегменту, тим легше він переміщується за даної температурі й, отже, тим менше час релаксації τ. З ростом полярності й потенційного бар’єра обертання в макромолекулах збільшується час релаксації τ. Весь комплекс механічних властивостей полімеру визначається співвідношенням τ/t. Це співвідношення називається критерієм Дебори й позначається D= τ/t. Чим менше D, тим швидше релаксує система, тим вона податливіша. При малих значеннях D полімери виявляють властивості рідин, а при більших − властивості твердих тіл. Наявність границі розподілу полімер−тверде тіло впливає на окремі релаксаційні механізми. Однак, зміни часів релаксаційних процесів, пов’язаних з рухом бокових груп і сегментів ланцюгів, невеликі й не можуть пояснити різкого підвищення Т с полімерів, що перебувають у тонких шарах на поверхні полімеру. Це пов’язане з тим, що Т с визначається рухливістю як дуже малих сегментів ланцюгів, так і більших структурних елементів, аж до рухливості надмолекулярних структур. Отже, повинна існувати група релаксаційних процесів, обумовлених, головним чином, рухливістю структурних елементів і для цієї групи повинно спостерігатися помітне зростання середніх часів релаксації внаслідок взаємодії з поверхнею. Відомо, що ізотермічний стиск може бути описаний за допомогою рівняння
де V – об’єм у момент часу t; V∞ - рівноважний об’єм; τср – середній час релаксації. Для t = τср
На цій підставі середні часи релаксації можуть бути знайдені графічно (рис. 2.2).
За даної температури середні часи релаксації процесу ізотермічного стиснення підвищуються для полімеру, що перебуває на поверхні твердих часток. Це доволі суттєво з погляду вибору умов переробки наповнених полімерів, тому що цілком очевидно, що для одержання наповненого полімеру з оптимальними властивостями режим формування (температура, час, тиск) повинен відрізнятися від режиму формування того ж полімеру без наповнювача. Унаслідок обмеження рухливості макромолекул поверхнею наповнювача підвищується температура склування в поверхневому шарі. Тс збільшується незалежно від того, чи є таке обмеження наслідком енергетичної взаємодії з поверхнею, чи обумовлено геометричним ентропійним фактором. Цим збільшенням пояснюються більші часи релаксації для поверхневих шарів у порівнянні з часами релаксації в об’ємі при однакових температурах. Однак, зменшення щільності упаковки полімеру в поверхневому шарі, і як наслідок збільшення вільного об’єму, при температурах, рівновіддалених від Тс, полегшує протікання релаксаційних процесів і знижує часи релаксації. Аналогічна картина спостерігається і за перетворення від лінійних полімерів у зшиті. Однак, у цьому випадку середні часи релаксації полімеру в поверхневих шарах значно ближче до середніх часів релаксації в об’ємі, чим у випадку лінійних полімерів. Ці явища можуть бути пояснені таким чином. Обмеження рухливості ланцюгів на поверхні розподілу може бути викликано двома причинами. Перша − це адсорбційна взаємодія макромолекул або надмолекулярних утворень із поверхнею (енергетична взаємодія). Вона накладає певні обмеження на рухливість ланцюгів і внаслідок цього – на конфігураційний набір макромолекул у поверхневому шарі. Інша причина може носити чисто ентропійний характер. Конфігураційний набір макромолекул поблизу границі поділу зменшений у порівнянні з таким в об’ємі з суто геометричних причин: на поверхні макромолекула не може прийняти ту ж кількість конформацій як в об’ємі. Це фактично еквівалентно підвищенню жорсткості макромолекули в поверхневому шарі й, отже, зниженню її рухливості. Тому у випадку помірної взаємодії макромолекул з поверхнею обмеження рухливості на границі поділу можуть носити ентропійний характер. Таке обмеження рухливості еквівалентно явищам механічного склування, що полягає у втраті ланцюгами здатності до зміни конформації при збіднінні їхнього конфігураційного набору. Таким чином, при розгляді обмеження рухливості полімерних ланцюгів поблизу границі поділу необхідно враховувати спільний вплив на це обмеження як енергетичного так й ентропійного фактора. Спільний їхній вплив приводить до того, що ланцюг у поверхневому шарі стає жорсткішим й, отже, механічні властивості такого шару повинні відрізнятися від властивостей полімеру в об’ємі. Слід зазначити, що зміна властивостей поверхневих шарів полімеру в порівнянні із властивостями в об’ємі визначається, головним чином, саме наявністю границі розподілу й мало залежить від молекулярної маси полімеру. Ефекти зміни Т с і часів релаксації для зшитих полімерів стають усе менш помітними зі збільшенням густоти просторової сітки. Поперечне зв’язування ланцюгів приводить до зниження рухливості, в першу чергу, довгих ділянок ланцюгів. Чим густіше просторова сітка, тим нижче рухливість. А з огляду на те, що саме рухливість довгих ділянок визначає часи релаксації і на поверхні, і в об’ємі, то для зшитих полімерів означені ефекти нівелюються. Ріст Т с на поверхні досягає всього 2−3 °С, як і у випадку зниження тільки сегментальної рухливості лінійних полімерів. Таким чином, зі збільшенням густоти просторової сітки й зменшенням внаслідок цього розмірів ділянок ланцюгів, що беруть участь у релаксаційному процесі, вплив поверхні на них зменшується. Цей вплив, однак, позначається на зміні щільності упаковки полімеру й на умовах виникнення сітки в ході хімічної реакції. Як правило, за температур, менших Т с, енергії активації процесів об’ємної релаксації вищі, ніж при більш високих температурах. Це пов’язане з тим, що нижче Т с можливий прояв рухливості значно менших структурних елементів, ніж за температур вище Т с. Нижче Т с спостерігається також деяке зростання енергій активації зі збільшенням вмісту наповнювача, тобто зі зменшенням товщини поверхневого шару. Вище температури склування енергія активації практично не залежить від товщини шару. Менші значення енергії активації в низькотемпературній області можуть також бути пов’язані з нещільністю упаковки, що прогресує в міру збільшення вмісту наповнювача, а більші значення у високотемпературній області пов’язані із проявом рухливості більших ділянок ланцюгів. Звертає на себе увагу той факт, що у зшитих полімерах енергії активації за температур нижче Т с трохи зростають із ростом вмісту твердих часток, але ці відмінності нівелюються зі збільшенням густини просторової сітки. У випадку застосування у якості наповнювачів волокон із синтетичних полімерів часи релаксації й енергії активації для даного полімеру мало залежать від типу поверхні. Це дозволяє зробити висновок про те, що в механізмі обмеження рухливості великих елементів ланцюгів поблизу границі розподілу найбільшу роль грають процеси, пов’язані зі збіднінням конфігураційного набору макромолекул, тобто природа полімеру, а не процеси енергетичної взаємодії між полімером і наповнювачем. Введення пластифікаторів у полімер знижує Т с, але мало впливає на енергію активації, тому що він не порушує зв’язки на границі розподілу фаз. Якби ефект був пов’язаний з енергетичною взаємодією, вплив пластифікатора був би більш вираженим. Той же висновок випливає з даних відносно часів релаксації пластифікованих полімерів.
|
||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-06; просмотров: 415; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.41 (0.009 с.) |

 ,
,
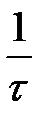 – коефіцієнт пропорційності, що залежить головним чином від структури й властивостей досліджуваної системи, і зворотний величіні часу релаксації. Після нескладного перетворення і інтегрування (від 0 до t і від σ0 до σ) отримуємо:
– коефіцієнт пропорційності, що залежить головним чином від структури й властивостей досліджуваної системи, і зворотний величіні часу релаксації. Після нескладного перетворення і інтегрування (від 0 до t і від σ0 до σ) отримуємо: ,
,
 , тобто час релаксації τ – це час, за який початкове напруження σ0 зменшиться в е раз (t – повний час встановлення рівноважного стану) Швидкість релаксації тим більше, чим менше τ. З іншого боку, τ тим менше, чим більше швидкість теплового руху сегментів, тобто τ зменшується з ростом температури. Залежність часу релаксації від температури виражають рівнянням Арреніуса–Ейринга–Френкеля:
, тобто час релаксації τ – це час, за який початкове напруження σ0 зменшиться в е раз (t – повний час встановлення рівноважного стану) Швидкість релаксації тим більше, чим менше τ. З іншого боку, τ тим менше, чим більше швидкість теплового руху сегментів, тобто τ зменшується з ростом температури. Залежність часу релаксації від температури виражають рівнянням Арреніуса–Ейринга–Френкеля:  ; або у логарифмічному вигляді −
; або у логарифмічному вигляді −  Тоді Е = R· tg α (Рис. 2.1.).
Тоді Е = R· tg α (Рис. 2.1.).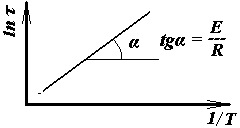
 ,
,
 .
.




