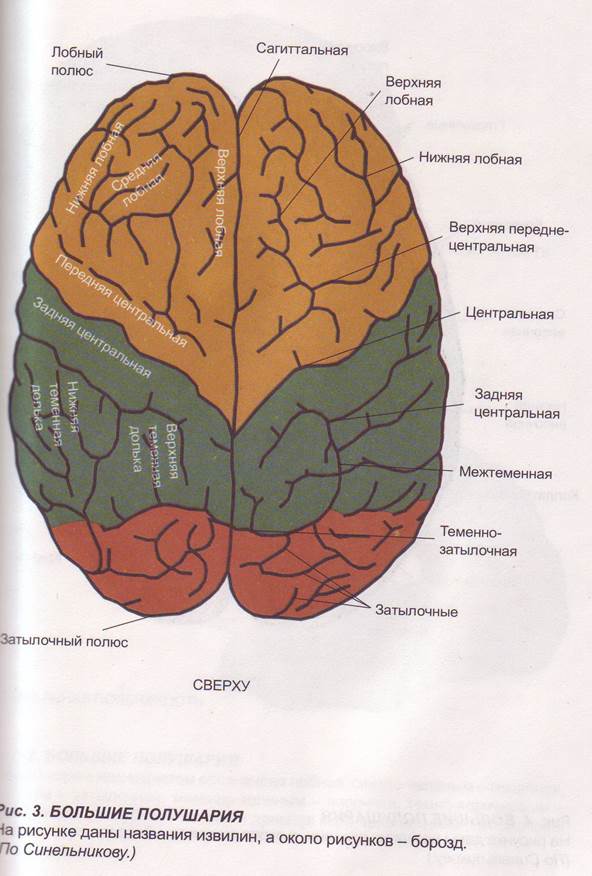
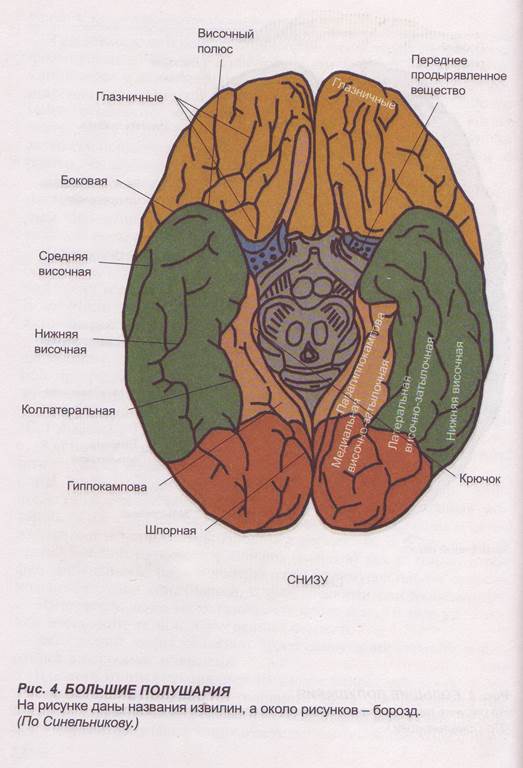
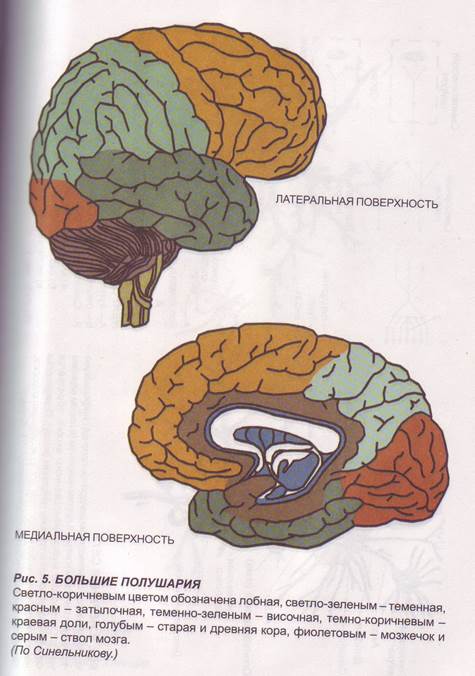
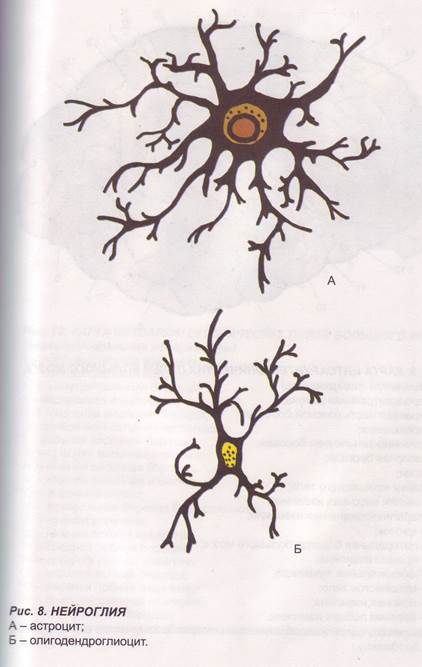
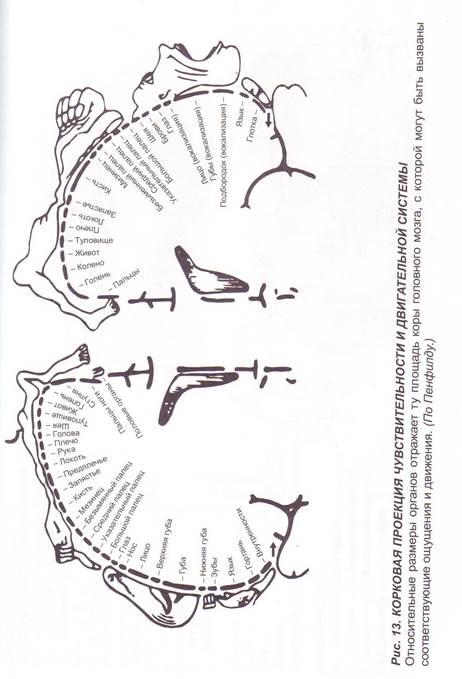
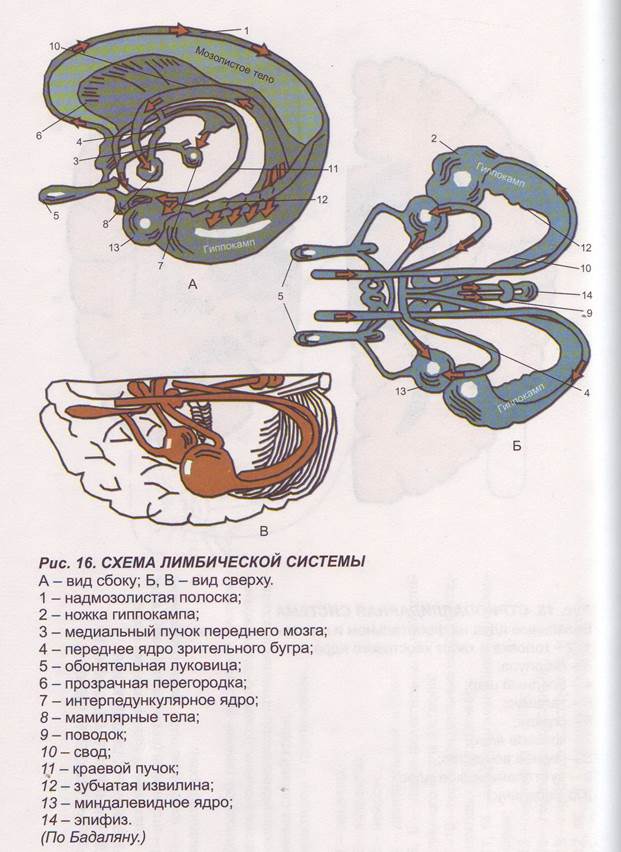
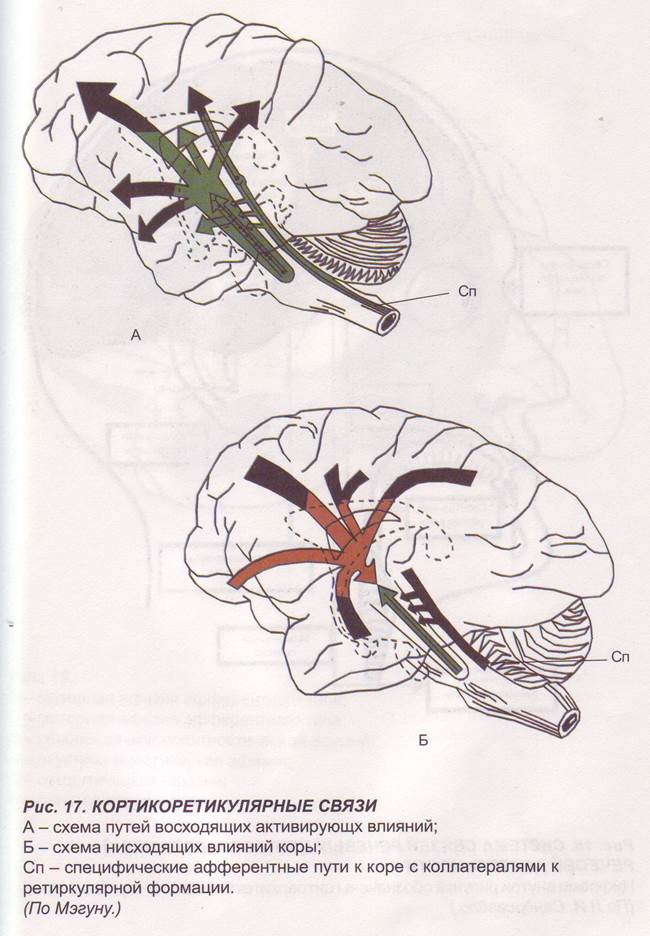
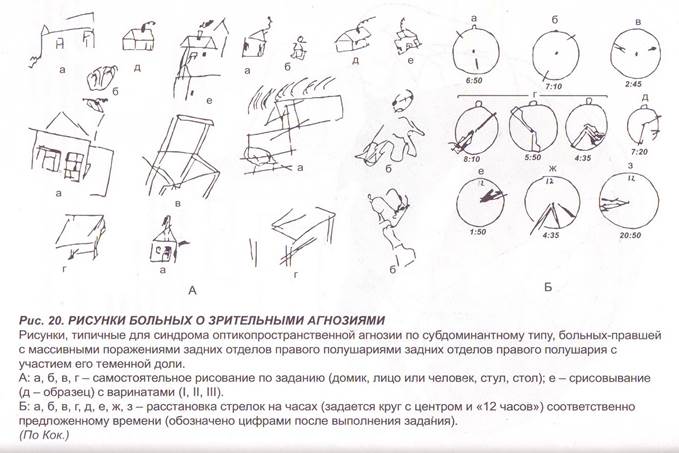
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Умственная отсталость, обусловленная другими уточненными причинамиСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Среди других уточненных причин, вызывающих умственную отсталость, наиболее распространенными и важными, по мнению авторов, являются заболевания, обусловленные: резус-несовместимостью крови матери и плода, дисгенетической и диспластической патологией мозга, поражением эндокринной системы, нарушением развития и изменением функций определенных тканей, а также нарушением обмена веществ и дисферментозами. Гемолитическая болезнь новорожденных — заболевание новорожденных вследствие резус-несовместимости крови матери и плода. Различают три клинических формы заболевания: 1) отечная, сопровождающаяся общими отеками тела, выраженной гидроцефалией, бледностью и истонченностью кожи; 2) анемическая, при которой выявляется резкое малокровие внутренних органов и мозга; 3) желтушная, характеризующаяся прогрессирующей желтушностью кожных покровов. При любой из форм гемолитической желтухи новорожденных отмечаются поражения нервной системы, которые проявляются в виде синдрома умственной отсталости, глухоты, гиперкинезов, параличей конечностей и др. Обменное переливание крови в виде курса лечения, проведенное своевременно, снижает тяжесть поражения нервной системы. К биогенетическим и диспластическим заболеваниям мозга относится ряд уродств развития головного мозга, развившихся, как правило, в результате различных инфекций или интоксикаций в перинатальном периоде. Это прежде всего: анэнцефалия, гидроанэнцефалия и микроцефалия (см. выше). Поражения эндокринной системы в большинстве случаев также приводят к различным проявлениям умственной отсталости. Функциональная связь между центральной нервной системой и эндокринными органами осуществляется уже в процессе эмбриогенеза. Несомненно, деятельность эндокринных органов матери отражается на развитии эндокринной системы эмбриона плода. Среди большой группы синдромов умственной отсталости эндокринного генеза наиболее часто встречаются заболевания, возникающие при поражениях гипоталамо-гипофизарного комплекса. Синдром Бабинского—Фрелиха (синдром адипозо-генитальной дистрофии) — заболевание нейроэндокринной системы, характеризующееся прогрессирующим ожирением, преимущественно в области живота, бедер, груди, недоразвитием наружных и внутренних половых органов, наличием импотенции у мужчин и аменореи — у женщин. Отсутствует оволосение на лобке и в подмышечных впадинах. На месте молочных желез — избыточное отложение жира, бледность сосков. Отмечается задержка роста, если синдром появляется в детском возрасте. У больных часто развивается клиника гипогликемической недостаточности из-за низкого уровня сахара в крови. Синдром адипозогенитальной дистрофии впервые описан Бабинским (J. Babinski) в 1900 г. и А. Фрелихом (A. Frelich) в 1901 г. при доброкачественных опухолях гипофиза. Агромегалия (акромегалоидный синдром, синдром П. Мари—Лери, пахиакрия) — заболевание, обусловленное гиперсекрецией сомототропного гормона передней доли гипофиза и характеризующееся чрезмерным ростом выступающих частей тела, увеличением внутренних органов и появлением некоторых эндокринных нарушений. Заболевание описано П. Мари (P. Marie) в 1886 г. Развитие заболевания постепенное и медленное. Синдром в одинаковой популяции встречается как у мужчин, так и у женщин, чаще среднего возраста. Вначале заболевания у больных появляются: => симптомы астенического синдрома (общая слабость, быстрая утомляемость, повышенная раздражительность, ухудшение сна); <=> приступообразные головные боли, преимущественно в височных областях; <=> снижение половой функции у мужчин и нарушение овариально-менструальной функции у женщин. Постепенно развивается типичная клиническая картина акромегалии: укрупнение черт лица, языка, ушей, кистей, стоп, размеров головы, увеличение надбровных и скуловых дуг, челюстей с нарушением прикуса, искривление позвоночника, огрубение голоса, дизартрия, морщинистость кожи лица, повышение потоотделения, лабильность пульса и артериального давления (А/Д.), нарушение зрения. У мужчин отмечается увеличение грудных желез (гинекомастия). В случаях развития заболевания в детском и подростковом возрасте наблюдается увеличение роста (гигантизм). Вся вышеописанная клиника сопровождается нарастающим симптомокомплексом интеллектуальной недостаточности. Гигантизм. Заболевание возникает в период роста, чаще в подростковом, пубертатном возрасте и характеризуется чрезмерным ростом тела и конечностей (190 см у женщин и 200 см у мужчин). На фоне усиленного роста туловища отмечаются относительно малые размеры головы, удлинение конечностей и увеличение размеров внутренних органов. Все эти изменения сопровождаются резким снижением интеллекта, эмоциональной и психической инфантильностью. Синдром Иценко—Кушинга (гипокортицизм, надпочечниковый синдром) — синдром, обусловленный гиперфункцией коры надпочечников с повышенным выделением глюкокортикоидных гормонов как результат врожденного либо приобретенного нарушения в гипоталамо-гипофизарной системе (опухоль, воспалительный процесс и др.). Заболевание впервые описано Алпертом (Alpert) в 1910 г. и Н.М. Иценко в 1926 г. Подробно изучено в 1932 г. Кушингом (Н. Cuching). Клиническая картина характеризуется непропорциональным ожирением с отложением жировой клетчатки на лице, шее, верхней половине туловища. Лицо круглое, приобретает вид «полной луны», цианотично-красное, а область затылка напоминает «вид горба бизона». Кожа сухая, тонкая, истонченная, шелушащаяся с цианотично-мраморным рисунком, в области живота, ягодиц, бедер отмечаются своеобразные «полосы беременных», на конечностях «мраморность кожи», иногда подкожные кровоизлияния. Часто выявляется гиперпигментация кожи в области половых органов и сосков. У женщин наблюдается выпадение волос на голове, нарушение менструального цикла, вплоть до полного его исчезновения (аменорея). У мужчин — резкое снижение полового влечения, утрата вторичных половых признаков и потенции, у детей — гипогенитализм. Характерным симптомом синдрома Иценко—Кушинга является повышенное оволосение у женщин (появление усов, бороды, рост волос на груди, спине, конечностях) и отсутствие оволосения у мужчин. Выявляется резкое нарушение психики (астения, депрессия, психопатизация личности) и снижение интеллекта. Синдром несахарного диабета — заболевание, обусловленное пониженной выработкой антидиуретического гормона как результат врожденной аномалии либо приобретенной патологией нейроэндокринной системы. Клиническая картина характеризуется повышенной жаждой и полиурией (усиленное мочеиспускание) - до 10 л и более в сутки, сухостью кожи, снижением пото- и слюноотделения, а также половой функции. У детей отмечается задержка роста. Со стороны психической сферы Выявляется выраженная общая астения и снижение интеллекта. Гипоталамо-гипофизарная кахексия (болезнь Симмондса—Шихена—Глинского) — симптомокомплекс недостаточности передней доли гипофиза. Синдром описан польским паталогом Глинским (К. Glinski) в 1913 г., немецким паталогом Симмондсом (М. Simmonds) в 1914 г. и шотландским эндокринологом Шихеном (J. Sheehan) в 1937 г. Наблюдается чаще у женщин, нередко после родов. Отмечаются общая слабость, расстройство стула, прогрессирующее исхудание, преждевременное старение, атрофия мышц, исчезновение половых признаков с атрофией половых органов, аменореей, у мужчин — импотенция. Кахексия сочетается с выраженными психическими симптомами, депрессией, прогрессирующим нарушением высших мозговых функций, вплоть до полного маразма. Гипофизарная карликовость. Генетическое заболевание, характеризующееся низким ростом до 120—130 см у взрослых. Причем при рождении ребенка рост и длина его нормальные. Отставание в росте отмечается с 2—4 лет, сочетается с отставанием в развитии половых признаков. Со стороны высших мозговых функций отмечается инфантилизм психической и эмоциональной сферы. Синдром Лоренса—Муна—Бидля—Барде (адипозо-генитальный синдром с врожденными пороками развития) — симптомокомплекс врожденных аномалий развития с умственной отсталостью и выраженной дисфункцией гипоталамической области.
■=> легкая либо умеренная умственная отсталость; о нейроэндокринное ожирение (преимущественное отложение жира на бедрах, ягодицах, животе); => выраженный гипогенитализм; ■=> нарушения зрения; ■=> разнообразные аномалии черепа, конечностей, позвоночника, ребер и внутренних органов. Кроме знаний о многочисленных проявлениях умственной отсталости, обусловленных нарушением развития и изменением функций определенных тканей, будущим специалистам необходимы знания о клинических особенностях следующих синдромов и заболеваний. Остеопороз (синдром Альберса—Шенберга), характеризующийся прогрессирующим течением, клинически проявляющимся истончением и «мраморностью» костей, карликовым ростом, поражением печени и селезенки, анемией, слепотой (атрофия зрительных нервов), экзофтальмом, нарастающей умственной отсталостью. Туберозный склероз (болезнь Бурневилля) — наследственное прогрессирующее заболевание, характеризующееся тяжелой или глубокой умственной отсталостью и множественными опухолями различных органов систем (надпочечники, мышечная ткань, периферические нервы, нейроглия, сосуды и др.). Заболевание заканчивается полным маразмом и смертью от истощения или разрыва сосудов внутренних органов и мозга. Синдром впервые описан французским невропатологом Бурневиллем (D.M. Bourneville) в 1880 г. Вертикальная складчатость кожи. Основными клиническими проявлениями заболевания являются нарастающая умственная отсталость и своеобразная складчатость кожи головы, как бы напоминающая поверхность мозга. Заболевание часто сопровождается судорожными приступами и эпилептическими припадками, а также аномалиями глазных яблок. Данная патология чаще наблюдается у лиц мужского пола и начинается, как правило, в подростковом возрасте. Нейрофиброматоз (болезнь Реклингхаузена) — наследственное к заболевание, характеризующееся образованием множественных опухолей кожи, подкожной клетчатки, периферических нервов и основе данного синдрома лежат пять основных клинических пигментных пятен на различных участках кожи и слизистых оболочках. Впервые описан в 1882 г. немецким патологом Ф. Реклингхаузеном (von F.D. Recklinghausen) Основными клиническими проявлениями заболевания являются появления на коже, в подкожной клетчатке и по ходу нервных стволов опухолевых узлов, приводящих к нарушению различных видов чувствительности и движений в зоне иннервации пораженного нерва. При разрастании опухолевых образований в области спинного мозга и его корешков преобладают симптомы сдавления спинного мозга с параличами и парезами соответственно пораженным сегментам. Локализация опухолей в области шеи и дыхательных путей сопровождается нарастающим затруднением дыхания, а сдавление кровеносных и лимфатических сосудов — нарушением местного кровообращения и отеком тканей. Часто в патологический процесс вовлекается костная система, что приводит в конечном итоге, к патологическим переломам. Первые признаки заболевания обнаруживаются сразу после рождения или в детском возрасте, при этом отмечается отставание детей в физическом и умственном развитии. Клиника интеллектуальных нарушений проявляется в виде умеренной умственной отсталости. Синдром Луи—Бара — прогрессирующее заболевание, которое развивается в раннем детском возрасте и характеризуется различными неврологическими расстройствами, кожными и дистрофическими проявлениями, в сочетании с умеренной умственной отсталостью. Впервые синдром описан французским врачом Луи Баром (Louis Bar D.) в 1941г. Клиническая картина носит полиморфный характер и включает в себя следующие основные симптомы: ■=> нарушение координации движений; <=> шаткость при ходьбе; <=> скандированая, монотонная речь; <=> диффузная мышечная гипотония; ■=> веснушкоподобная сыпь на лице, придающая коже цвет «кофе с молоком»; => раннее поседение волос; <=> общая кахексия;
У больных с синдромом Луи Бара часто отмечаются развитие злокачественных опухолей и рецидивы хронических заболеваний. Умственная отсталость проявляется полным симптомокомплексом умеренной степени. Синдром Лоу—Терри—Мак—Лехиана (окулоцереброренальный синдром) — наследственное заболевание, характеризующееся поражением почек и глаз в сочетании с умственной отсталостью. Синдром впервые описан в 1952 г. американским врачом-педиатром Лоу (U. Lowe) с соавторами. Клиника заболевания характеризуется умеренной либо тяжелой умственной отсталостью, развитием у больного катаракты, глаукомы и ограничением подвижности глазных яблок. Патология почек проявляется в виде отеков конечностей, болей в поясничной области, нарушений мочеиспускания и других симптомов, в зависимости от степени канальцевой недостаточности почек. На фоне вышеперечисленных симптомов у больных часто выявляются эндокринные нарушения в виде карликового роста, остеопороза трубчатых костей, седловиднообразного носа и др. Синдром Марфана — синдром, обусловленный наследственным пороком развития соединительной ткани и характеризующийся поражением опорно-двигательного аппарата, внутренних органов, глаз и интеллектуальной недостаточностью. Заболевание описано впервые в 1896 г. французским врачом-педиатром А. Марфаном (B.J.A. Marfan) Клиническая картина проявляется резко выраженной астенической конституцией тела (килевидная или воронкообразная грудная клетка с широкими межреберными промежутками и тонкими длинными ребрами), «птичьим» выражением лица (узкий череп, срезанный подбородок, близко посаженные глаза), тонкими ушными раковинами, синюшным оттенком склер глаз, подвывихом или вывихом хрусталика, общей мышечной гипотонией, недоразвитием подкожной клетчатки, слаборазвитой мышечной тканью, разболтанностью суставов, удлиненными кистями и стопами с тонкими «паукообразными» пальцами, вегетативно-сосудистыми нарушениями (повышенная потливость, мраморность кожи, акроцианоз), пороками сердца. Интеллектуальная недостаточность носит характер легкой или умеренной умственной отсталости. Прогноз в связи с возможными осложнениями во всех случаях неблагоприятный. Гипертелоризм. В основе данного синдрома лежит неправильное формирование черепа в эмбриональном периоде, в связи с чем клинически у детей наблюдается деформация черепа в виде расширения переносицы и широко поставленных глазниц. Одновременно отмечаются врожденные аномалии скелета кистей. Интеллектуальные нарушения носят вторичный характер как результат задержки психического развития. Заболевание носит семейный характер, чаще болеют девочки. Челюстно-лицевой дизостоз. Заболевание характеризуется деформацией черепа, недоразвитием верхней челюсти, преждевременным заращением швов черепа, рядом анатомических аномалий и тяжелой умственной отсталостью. Прогрессирующий окостеневающий миозит — наследственное заболевание, характеризующееся постепенным уплотнением мышечной ткани всего организма и интеллектуальной недостаточностью. В конечном итоге у больных развиваются почти полная обездвиженность, затруднение жевания и дыхания. Синдром Пелицеуса—Мерцбахера (хроническая форма семейного диффузного склероза) — заболевание, обусловленное наследственной прогрессирующей лейкодистрофией у грудных детей с признаками нарушения липоидного обмена. Впервые описан немецкими невропатологами Ф. Пелицеусом (F. Pelizaeus) в 1885 г. и Л. Мерцбахером (L. Merzbacher) в 1908 г. Клиническая картина данной патологии проявляется уже в грудном возрасте, когда у младенца появляются дрожание головы, нистагм (непроизвольные подергивания глазными яблоками), диффузная мышечная слабость, постепенно присоединяется общая скованность и спастические парезы конечностей с повышением сухожильных и исчезновением брюшных рефлексов. В более поздней стадии болезни развивается амовроз (слепота) как результат атрофии зрительных нервов и контрактуры, преимущественно проксимальных отделов конечностей, с трофическими расстройствами. Интеллектуальная недостаточность проявляется в виде умственной отсталости легкой и умеренной степени. Заболевание носит медленно прогрессирующий характер и поражает в основном мальчиков. В основе многих прогридиентных врожденных заболеваний лежит органическое поражение головного мозга, обусловленное тотальным нарушением определенного вида обмена веществ, и прежде всего ферментов, участвующих в осуществлении нормального протекания тех или иных биохимических процессов. К этой группе патологии относятся следующие синдромы..
Синдром Гурлера (гаргоилизм) — заболевание, характеризующееся накоплением мукополисахаридов (основные «цементирующие» вещества соединительной ткани — гиалуроновая и хондриотинсерная кислоты) в клетках головного мозга. Накопление этих кислот является результатом врожденного дефицита фермента гиалуронидазы, расщепляющего гиалуроновую кислоту. Впервые синдром описан Гурлером (J. Hurler) в 1920 г. Основными клиническими симптомами заболевания являются: низкий рост, широкие черты лица, деформированные череп, грудная клетка, кисти рук, крупные толстые губы, огромный язык, не помещающийся в полости рта, сглаженная переносица, укороченная шея, толстые пальцы, вздутие живота вследствие поражения селезенки. Характерным своеобразным симптомом является «вывернутые вперед широкие ноздри», придающие лицу «выражение лица человека, выплескивающего воду (гаргоилизм)». Умственная отсталость колеблется от умеренной до тяжелой степени. Миоклонус-эпилепсия (синдром Унферрихта—Лундборга) — наследственное заболевание, впервые описанное немецким терапевтом Унферрихтом (Н. Unverricht) в 1891 г. и шведским невропатологом Лундборгом (Н. Lundborg) в 1903 г. Русский невропатолог В.М. Бехтерев в 1897 г. описал клинику данного синдрома под названием «хореическая эпилепсия». Клиническая картина заболевания разделяется на 3 стадии. 1 стадия — эпилепто-тетаническая — характеризуется: ■=> эпилептическими приступами, сопровождающимися потерей сознания, прикусом языка, непроизвольным мочеиспусканием, тоническими и клоническими судорогами в конечностях преимущественно в ночное время; ■=> психическими расстройствами (депрессия либо повышенная возбудимость, агрессия); ■=> умственной отсталостью (прогрессирующей) — от симптомов легкой до умеренной степени. 2 стадия — миоклонически-эпилептическая. При этой стадии заболевания отмечается прогрессирование клинической симптоматики, которая проявляется в виде увеличения частоты судорожных приступов, распада личности и тяжелой умственной отсталости. 3 стадия — терминальная (конечная). При этой стадии заболевания на фоне эпилептических приступов у больных появляются практически постоянные миоклонические судорожные подергивания, напоминающие хореические (непроизвольные подергивания в различных мышечных группах), но отличающиеся большей интенсивностью, особенно при судорогах мышц головы и рук. Миоклонии усиливаются при активных движениях и даже незначительном перевозбуждении и волнении. Отмечается быстрый прогресс психических и интеллектуальных расстройств, вплоть до полного распада личности и глубокой умственной отсталости. Синдром Нимана—Пика — наследственное заболевание, обусловленное нарушением липидного обмена и накоплением в нервной ткани ганглиозида, способствующего развитию глубокой умственной отсталости. Впервые синдром описан немецким педиатром Ниманом (A. Niemann) в 1914 г. и немецким патологом Пиком (Pick L.) в 1926 г. Клиническая картина характеризуется монголоидным видом лица, желто-коричневым цветом кожи, прогрессирующим похуданием, снижением слуха и зрения, умственным слабоумием различной степени тяжести, вплоть до глубокой. При данной патологии часто отмечается сочетание вышеописанных симптомов с различными аномалиями (истончение кожи, остеопороз и др.). Прогноз неблагоприятный. Летальный исход наступает приблизительно в 5—8-летнем возрасте. Болезнь Гоше (керазиновый ретикулоэндотелиоз). Заболевание сходно с болезнью Нимана—Пика и является наследственной энзимопатией, обусловливающей постепенное накопление в ретикулоэндотелиальных клетках так называемых цереброзидов (галактозоцереброзидов, гликоцереброзидов, керазина). Сопровождается прогрессирующим слабоумием, гепатоспленомегалией, поражением костной системы, угнетением кроветворения, пигментацией кожи и роговицы. Заболевание, начинающееся в самом раннем возрасте, течет весьма злокачественно, так как, помимо слабоумия, отмечают и органические неврологические симптомы, а именно: псевдобульбарные явления, косоглазие, спастические парезы, в конечных стадиях — децереброционную ригидность. Мозг детей страдает из-за прогрессирующей дегенерации ганглиозных клеток. (В.В. Русских, 1969). Заболевание впервые описано французским дерматологом Гоше (Е. Gaucher) в 1982 г. Амавроатическая идиотия Тея—Сакса — наследственное, семейное заболевание, характеризующееся слепотой (амавроз) в сочетании с глубокой умственной отсталостью. Впервые описано английским офтальмологом Теем В. (W. Тау) в 1881 г. и американским невропатологом Б. Саксом, (В. Sachs) в 1898 г. Первые клинические симптомы в виде снижения зрения, вплоть до полной слепоты, развиваются к концу первого года жизни. Нарушение зрения является результатом прогрессирующей атрофии зрительного нерва. Постепенно присоединяются двигательные нарушения в виде центральных парезов и параличей конечностей. Часто наблюдаются эпилептиформные приступы с тоническими и клоническими судорогами конечностей, а также различные гиперкинезы. В течение от 6 месяцев до 1 года развивается клиника глубокой умственной отсталости. Продолжительность жизни больных с данной патологией 3—4 года. Синдром Доллингера-Бильшовского (амавротическая поздняя идиотия) — наследственное заболевание, которое характеризуется нарушением жирового обмена и клинически проявляется глубокой умственной отсталостью в сочетании со слепотой. Заболевание описано чешскими врачами Янским (Jansky) и Шобом (Schob) в 1910 г., немецкими невропатологами Бильшовским (М. Bielschovsky) в 1914 г. и Доллингером (A. Dollinger) в 1919 г., поэтому более правильное название данного синдрома — синдром Янского-Шоба-Бильшовского—Доллингера. Первые признаки болезни отмечаются приблизительно в трехлетнем возрасте, когда у больных выявляются признаки остановки психического развития и утраты уже приобретенных навыков речи и ходьбы, вплоть до глубокой степени умственной отсталости (в течение 1-2 лет). Постепенно развивается слепота на оба глаза в результате очаговой атрофии сетчатки, а также различные неврологические проявления (нарушения координации движений, приступы клонических судорог в конечностях, бульбарный паралич). Продолжительность заболевания — 3-5 лет. Синдром Ван—Богарта—Шерера—Эпштейна (синдром липидоза) — заболевание, характеризующееся нарушением жирового, холестеринового обмена. Впервые описан в 1937 г. бельгийскими невропатологами Ван-Богартом (Van-Bogaert) и Шерером (Н. Scherer) и австрийским биохимиком Эпштейном (Е. Epstein). Клиника заболевания проявляется прогрессирующей, умственной отсталостью от легкой до тяжелой степеней, изменением кожных покровов (сухость, морщинистость, множественные внутрикожные и подкожные кровоизлияния), ломкостью волос, выпадением зубов, остеопорозом, искривлениями позвоночника, катарактой, выраженным гипогенитализмом и неврологическими расстройствами в виде развития мозжечковой атаксии и бульбарного паралича (нарушение глотания, дизартрия). Синдром Бассена—Корнцвейга — наследственное заболевание, характеризующееся поражением мозжечка, спинного мозга, дегенерацией сетчатки глаз, снижением уровня холестерина в крови и умственной отсталостью. Впервые описан американскими врачами Бассеном (F. Bassen) и Корнцвейгом (A. Kornzweig) в 1950 г. Клиническая картина синдрома проявляется прогрессирующей атаксией (шаткость при ходьбе), нарушением координации движений, мышечно-суставного чувства и чувства пассивных движений, слабостью мышц проксимальных отделов конечностей, отеком дисков зрительных нервов с последующей дегенерацией сетчатки и умственной отсталостью умеренной или тяжелой степени. Синдром Леша—Нихема — врожденное заболевание, характеризующееся умственной отсталостью, аутоагрессией и нарушением обмена пуринов. Впервые синдром описан американскими врачами Лешом (М. Lesch) Нихемом (W.L. Nyham) в 1964 г. Первые симптомы заболевания появляются к 9- 10 месяцу после рождения, когда ребенок отказывается от пищи, уменьшается его моторная, двигательная активность. Постепенно присоединяются гиперкинезы и миоклонии в различных мышечных группах, которые в течение 1 года становятся менее выраженными из-за развития гипертонии мышц. В течение последующих 1—2 лет у ребенка возникают аутоагрессивные стремления (покусывание пальцев, рук, губ, языка). Нарушение обмена пуринов (повышенное содержание мочевой кислоты в моче и крови) проявляется в виде подагрических воспалений суставов и образования камней в почках, а также токсического влияния на различные отделы головного мозга, приводящие к демиелинизации (разрушение миелина) в полушариях головного мозга и мозжечка. Умственная отсталость характеризуется клинической картиной тяжелой либо глубокой степени тяжести. Синдром Зейтелбергера (спастическая амавротическая идиотия) — наследственное заболевание, обусловленное прогрессирующей липоидной (жировой) дегенерацией в центральной нервной системе. Впервые описан австрийским невропатологом Зейтелбергером (F. Seitelberger) в 1952 г. Клиника заболевания характеризуется выраженной задержкой психомоторного развития, приводящей к глубокой умственной отсталости, слепотой (амавроз) и глухотой. Постепенно присоединяются стволовые нарушения в виде нарушения глотания, параличей мышц лица, языка и жевательной мускулатуры, а также периферических параличей мышц шеи и затылка. Синдром Вильсона (гепато-лентикулярная дегенерация) — наследственное заболевание, характеризующееся поражением подкорковых узлов (чечевидное ядро) и паренхимы печени. Впервые подробно описано английским невропатологом Вильсоном, (S.A.K. Wilson) в 1912 г. В основе заболевания лежит увеличение содержания меди в печени и в головном мозге, приблизительно в 10 раз по сравнению с нормой. Клиника заболевания проявляется гипокинетическо-гипертоническим синдромом в сочетании с гиперкинезами, прогрессирующей умственной отсталостью и циррозом печени. Основными симптомами заболевания являются: маскообразное лицо, замедленность движений, общая скованность, иногда непроизвольные хореические подергивания в различных мышечных группах, желтушность склер, резкое увеличение печени. Умственная отсталость колеблется от легкой до умеренной степени. Вследствие различных инфекционных факторов и интоксикаций синдром Вильсона может носить и приобретенный характер с развитием, в конечном итоге, дементного синдрома. Болезнь Ганда—Шюллера—Христиана. «Семейный системный дисферментоз», прогрессирующее заболевание соединительной ткани, сопровождающееся разрушением мембранозных костей, вторичным поражением мозга, приводящим к слабоумию, экзофтальму, параличам глазных мышц и ряду других неврологических симптомов. Мозг может быть поражен процессом первично, что проявляется в патоморфологических изменениях серого бугра, белого вещества мозга. Гранулемные ксантомные очаги (характерные для заболевания) могут располагаться в оболочках мозга и сдавливать черепно-мозговые нервы, а также создавать выбухание и дефекты черепа. Локализация их на основании черепа вызывает несахарный диабет и синдром Фрейлиха, иногда карликовость. Рентгенологически определяются дефекты в костях черепа, таза, реже —-в длинных костях. Поражение височных костей может вызвать глухоту. Заболевание сопровождается кожными изменениями — желтоватыми ксантомами, которые можно использовать для биопсии. Болезнь нередко начинается в детском возрасте, течет медленно; для диагноза, помимо биопсии, используются данные о триаде: экзофтальме, несахарном диабете и дефектах в черепе» (В.В. Русских, 1969). Болезнь Летерер—Зиве. «Эта болезнь близка по природе к болезни Ганда—Шюллера—Христиана; обозначают как лейкемический ретикулез без изменения состава крови. Начинается в раннем возрасте. Приводит к деменции и общему истощению. Клиническая картина складывается из образования опухолевых очагов в черепе, позвоночнике, костях таза и конечностях» (В.В. Русских, 1969). Гликогеноз (гликогенная болезнь) наследственный дисферментоз, обусловленный недостаточностью ферментов, превращающих гликоген в сахар, и характеризующийся скоплением гликогена в клетках внутренних органов, мышцах и нейрологией. Заболевание, как правило, носит семейный характер и встречается у нескольких детей в одной семье. Впервые больной гликогенозом был описан в 1910 г. Леревуайе (Lerevouillet). В 1928 г. Ван-Кревельд (S.van Creveld) описал клиническую картину, а в 1929 г. Гирке (E.von Gierke) — патологоанатомическую картину этого заболевания, установив при нем накопление гликогена в печени и почках. Клиника заболевания проявляется уже в раннем возрасте и характеризуется тошнотой и рвотой, ухудшением аппетита, судорожными явлениями, вызванными увеличением количества гликогена в крови. Постепенно у детей нарушается походка, развивается гипотония мышц, замедляется рост, увеличивается печень, изменяются размеры черепа и позвоночника. Одним из основных признаков заболевания является развитие прогрессирующего слабоумия в виде умственной отсталости вплоть до тяжелой, а иногда и глубокой степени. Галактоземия — наследственный дисферментоз, обусловленный нарушением обмена галактозы. Галактоза-моносахарид, обладающий токсичным действием и входящий в состав лактозы (главный углевод грудного молока). В норме под влиянием фермента галактозы происходит расщепление галактозы, что приводит к полному распаду молочного сахара (лактозы) и усвоению организмом молока и молочных продуктов. Нарушение обмена галактозы у человека приводит к развитию тяжелых заболеваний, а кормление детей молочной пищей приводит к задержке умственного развития, а зачастую и к летальному исходу. Клиника заболевания проявляется уже в первые дни после рождения, как только ребенок начинает получать молоко с повышенным содержанием галактозы. Падение веса, желтуха, асцит (накопление жидкости в брюшной полости), диспептические расстройства являются основными симптомами начальных проявлений галактоземии. В дальнейшем развиваются катаракта и проявления умеренной либо тяжелой умственной отсталости. Ранняя диагностика и перевод ребенка на безмолочную диету в большинстве случаев позволяют избежать тяжелых умственных нарушений. Гистидинемия — наследственное заболевание, обусловленное нарушением обмена гистидина и характеризующееся умственной отсталостью различной степени, нарушением речи (моторная алалия) и рядом неврологических проявлений (атаксия, интенционное дрожание, судорожный синдром). Впервые описано в 1961 г. Гадими (Н. Ghadimi). Гомоцистинурия — наследственное заболевание, обусловленное нарушением обмена метионина и характеризующееся поражением соединительной ткани, а также нервной, костной, мышечной и сердечно-сосудистой систем. Впервые описано в 1962 г. Карсоном и Нейллом (N.A.J. Carson, D.W. Neill). Клиническая картина сходна с клиникой синдрома Марфана, однако повышенное содержание метионина в крови — до 30мг% (N—0,4—0,5мг%)? характерная «утиная походка» (походка Чаплина), а также развитие тяжелых церебральных нарушений (гемипарезы, атаксия и др.), обусловленных нарушением обмена аминокислот, и частые сосудистые «катастрофы» (тромбозы, эмболии) дают основание выделить данную патологию в число заболеваний с тяжелым течением и зачастую неблагоприятным исходом.
|
||||
|
|
Последнее изменение этой страницы: 2016-06-23; просмотров: 442; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.147.51.72 (0.022 с.) |

 Впервые заболевание описано английскими офтальмологами Лоренсом (J. Lourence) и Муном (R. Moon) в 1866 г., подробная характеристика дана австрийским физиологом Бидлем (A. Biedli) в 1922 г. и чешским терапевтом Барде (G. Bardet) в 1920 г.
Впервые заболевание описано английскими офтальмологами Лоренсом (J. Lourence) и Муном (R. Moon) в 1866 г., подробная характеристика дана австрийским физиологом Бидлем (A. Biedli) в 1922 г. и чешским терапевтом Барде (G. Bardet) в 1920 г. В основе данного синдрома лежат пять основных клинических проявлений:
В основе данного синдрома лежат пять основных клинических проявлений:
 => низкий рост.
=> низкий рост.