Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Влияние концентрации фермента на скорость ферментативной реакции.Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Концентрация фермента оказывает существенное влияние на скорость ферментативной реакции. При насыщающей концентрации субстрата, обеспечивающей Vmax, начальная скорость ферментативной реакции будет, в первую очередь, зависеть от концентрации фермента. Эта зависимость прямо пропорциональная, что свидетельствует о том, что начальная скорость является мерой количества фермента. Графически это представлено на рис. 8.5.
Влияние температуры на активность ферментов. Общий вид кривой, характеризующей влияние температуры на активность фермента, можно представить в виде графика, изображенного на рис. 8.6. Оптимальная температура, при которой наблюдается максимальная активность, для большинства ферментов находится в пределах 37-50 0C, но некоторые ферменты имеют температурный оптимум за пределами этой зоны.
Влияние температуры на активность фермента, которое может быть легко изучено экспериментально, имеет очень сложный характер, так как обусловлено целым рядом факторов, а именно: - влиянием температуры на скорость расщепления комплекса ES на свободный фермент и продукт реакции, т. е. на константу скорости реакции - влиянием температуры на сродство фермента к субстрату, то есть на константы k+1 и k _,; - влиянием на теплоту ионизации, а, следовательно, на процессы ионизации всех компонентов реакции: самого фермента, субстрата, промежуточных и конечных продуктов реакции; - влиянием на образование таких соединений, как "фермент-активатор" или "фермент-ингибитор"; - влиянием на процесс денатурации ферментного белка. Известное уравнение Аррениуса, характеризующее влияние температуры на скорость химической реакции, может быть приложено к левой части температурной кривой (см. рис. 8.6):
=
где k - константа скорости реакции; T - абсолютная температура, °К; E - энергия активации; R - универсальная газовая постоянная. Изменение скорости ферментативной реакции при повышении температуры измеряется температурным коэффициентом Q10, который показывает, во сколько раз ускоряется данная реакция при повышении температуры на десять градусов. Можно преобразовать уравнение Аррениуса, подставив в него коэффициент Q10:
E =
Это уравнение дает возможность определить энергию активации путем определения значений Q10 для данной ферментативной реакции. Для обычных химических реакций Q10= 2-3, для ферментативных реакций (левая часть температурной кривой) Q10= 1-2, причем значение Q10= 1 характерно для температур, близких к оптимальным. Правая часть температурной кривой показывает резкое снижение скорости ферментативной реакции при температурах, превышающих оптимальную. И это зависит, в первую очередь, от денатурации ферментного белка. Поэтому очень важным показателем, характеризующим отношение фермента к температуре, является его термостабильность. Термостабильность фермента складывается как бы из двух критериев: величины температуры и времени ее воздействия на фермент. Кроме того, на термостабильность различных ферментов могут оказывать влияние и такие факторы, как рН среды, ее солевой состав, защитное действие субстрата. Влияние рН на активность ферментов. Для каждого фермента характерна определенная узкая область значений рН, при которой он проявляет максимальную активность. Форма кривых, описывающих зависимость активности фермента от рН, отражает способность важных для данного фермента протон-донорных или протон-акцепторных групп в активном центре фермента переходить в состояние с требуемой степенью ионизации при определенных значениях рН (см. рис. 8.7).
Кроме влияния рН на состояние ионизации активного центра фермента, ход представленных кривых будет зависеть и от других факторов. В частности, изменение рН среды изменяет состояние ионизации субстрата (если это заряженное вещество), комплексов ES и EP, в некоторых, например, окислительно-восстановительных реакциях, ионы H+ сами могут принимать участие в реакции; помимо этого, скорость денатурации ферментативного белка зависит от рН. При экспериментальном изучении активности фермента от рН следует помнить, что рH-оптимум зависит от состава среды (от природы используемого буфера); оптимумы рН прямой и обратной реакции могут быть совершенно различными; при действии одного и того же фермента на различные субстраты рН-оптимумы также могут быть различными. Кроме понятия оптимума рН, очень важным является понятие рН-стабильности. Это тот диапазон рН, при котором фермент или ферментативный препарат сохраняет свою активность в течение определенного периода времени. рН-Стабильность также зависит от ряда факторов, среди которых, кроме уже названных, форма ферментного препарата, степень его очистки и др.
Все выше сказанное позволяет утверждать, что варьируя температурный режим и изменяя рН, можно в какой-то мере регулировать каталитическую активность фермента. Влияние активаторов и ингибиторов. Активаторами называют вещества, которые повышают активность ферментов. Хорошим примером таких соединений являются аминокислота цистеин и восстановленный глутатион, содержащие свободную SH-группу. Их активирующее действие заключается в том, что они восстанавливают дисульфидные связи с образованием SH-групп, необходимых для проявления каталитической активности тиоловых ферментов. Кроме того, некоторые ферменты активируются металлами, которые либо участвуют в построении активного центра, либо стабилизируют пространственную конформацию ферментного белка и тем самым обеспечивают проявление каталитических функций. Ингибиторами называют вещества, специфически снижающие активность ферментов. Снижение или полная потеря активности ферментов могут быть вызваны разного рода денатурирующими воздействиями, в этом случае правильнее употреблять термин "инактивация" фермента. Механизм действия ингибиторов может быть самым разнообразным: - ингибитор взаимодействует с апоферментом, при этом возможны такие варианты, как связывание функциональных групп белка, изменение третичной и четвертичной структуры апофермента, специфическое связывание с определенным участком апофермента, неспецифическая адсорбция на белке; - ингибитор образует комплекс с субстратом; - ингибитор связывает кофермент; - ингибитор связывает активатор; - ингибитор связывает кофактор. Чаще всего ингибитор взаимодействует с ферментом, образуя комплекс. Это можно выразить следующим уравнением:
Константа диссоциации комплекса фермент-ингибитор (или константа ингибирования) Ki.определяется выражением: ki =
=
Кi прямо пропорциональна концентрации фермента и ингибитора и обратно пропорциональна концентрации комплекса фермент-ингибитор. Существует ингибирование двух основных типов: необратимое и обратимое. Теоретически, в случае необратимого ингибирования k-1 = 0, то есть комплекс EI настолько прочен, что совершенно не диссоциирует. Но для большинства необратимых ингибиторов величина k-1 _, хотя и очень мала, но не равна нулю. Обратимое ингибирование, в свою очередь, бывает конкурентным и неконкурентным. Конкурентный ингибитор конкурирует с субстратом на основе структурного сходства, связываясь с активным центром фермента с образованием неактивного комплекса фермент-ингибитор. Отличительная особенность конкурентного ингибирования состоит в том, что его можно устранить или ослабить, повысив концентрацию субстрата. Конкурентный ингибитор снижает сродство фермента к субстрату, следовательно, величина Кm в присутствии конкурентного ингибитора увеличивается.
Графически это выглядит так, как изображено на рис. 8.8
При неконкурентном торможении ингибитор связывается не с активным центром фермента (то есть не там, где присоединяется субстрат), а с другим участком молекулы фермента. Очевидно, что в этом случае ингибитор не оказывает влияние на величину константы Михаэлиса Кт, но будет снижать максимальную скорость реакции - Vmax Графически это можно изобразить следующим образом (см. рис. 8.9):
Одними из самых распространенных неконкурентных ингибиторов являются аллостерические ингибиторы. Присоединяясь не к активному, а к другому, так называемому аллостерическому центру молекулы фермента, ингибитор вызывает конформационные изменения в структуре активного центра, вследствие чего становится невозможным образование комплекса фермент- субстрат. Изучение взаимодействия ферментов с ингибиторами и активаторами ферментов позволяет получать ценные сведения о субстратной специфичности ферментов, природе функциональных групп активного центра, механизмах каталитической активности. Так как в качестве ингибиторов могут выступать конечные продукты реакции, различные промежуточные продукты метаболизма, нет сомнения в той огромной роли, которую выполняют ингибиторы в регуляции ферментативной активности. Это подтверждает и факт широкого распространения ингибиторов белковой природы (см. гл. 2). Кроме того, по принципу специфического ингибирования действуют многие лекарственные препараты, антибиотики, токсичные вещества, антиалиментарные факторы питания. Специфические ингибиторы, встречающиеся в пищевом сырье и пищевых продуктах, присутствуют в качестве составляющих как в традиционных рецептурах, так и в сложных композиционных составах новых, модифицированных продуктов питания. Поэтому нельзя не учитывать их влияние на активность отдельных ферментов и на биохимические процессы в целом, протекающие при хранении и переработке пищевого сырья. Все это лишний раз говорит о множестве сложных проблем, которые встречаются в экспериментальной работе с ферментами и использовании ферментных препаратов на практике. 281:: 282:: 283:: 284:: 285:: 286:: 287:: 288:: 289:: 290:: 291:: 292:: 293:: 294:: 295:: Содержание 295:: 296:: 297:: 298:: Содержание 8.2. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА Катализируемая химическая реакция представляет собой тот специфический признак, по которому один фермент отличается от другого. Поэтому естественно и логично, что классификация и номенклатура ферментов основывается на этом принципе. Современная классификация ферментов разработана специальной Комиссией Международного Биохимического Союза и изложена в книге "Номенклатура ферментов", которая вышла в русском переводе в 1979 г.
В основе классификации лежат три положения: а) все ферменты делятся на 6 классов по типу катализируемой реакции; б) каждый фермент получает систематическое название, включающее название субстрата, тип катализируемой реакции, и окончание "аза"; кроме того, Комиссией были сохранены и узаконены тривиальные названия. Таким образом, возникла двойная система наименования ферментов; в) каждому ферменту присваивается четырехзначный шифр (код). Первое число указывает класс ферментов, второе — подкласс, третье — подподкласс, четвертое — порядковый номер фермента в подподклассе. Например, алкогольдегидрогеназа (Н.Ф.1.1.1.1): первая цифра — 1 — означает класс оксидоредуктаз, вторая цифра — 1 — подкласс дегидрогеназ (действует на СН — ОН-группу доноров), третья цифра — 1 — подподкласс анаэробные дегидрогеназы (акцептором служит НАД+ или НАДФ+), четвертая цифра — 1 — конкретный фермент алкогольдегидрогеназа. Или α-амилаза (Н.Ф.3.2.1.1): первая цифра — 3 — класс гидролаз, вторая цифра — 2 — подкласс карбогидраз, третья цифра — 1 — подподкласс полиаз, четвертая цифра — 1 — конкретный фермент α-амилаза. Современная международная классификация ферментов делит все ферменты на 6 основных классов: 1 класс — оксидоредуктазы — ферменты, катализирующие окислительно-восстановительные реакции (присоединение О2, отнятие и перенос Н2, перенос электронов); 2 класс — трансферазы — ферменты переноса. Катализируют перенос целых атомных группировок с одного соединения на другое (например, остатков моносахаридов, аминокислот, остатков фосфорной кислоты, метальных и аминных групп и т.д.); 3 класс — гидролазы — ферменты, катализирующие реакции гидролиза, то есть расщепления сложных органических соединений на более простые с участием воды. Эти реакции могут быть выражены следующим уравнением: RR1 + НОН → R — OH + R1 — H; 4 класс — лиазы — ферменты, катализирующие реакции негидролитического отщепления каких-либо групп от субстрата с образованием двойной связи или присоединение группировок по месту разрыва двойной связи (например, отщепление Н2О, СО2, NH3 и т.д.); 5 класс — изомеразы — ферменты, катализирующие реакции изомеризации, то есть внутримолекулярного переноса химических группировок и образование изомерных форм различных органических соединений; 6 класс — лигазы (синтетазы) — ферменты, катализирующие реакции синтеза, сопряженные с разрывом высокоэнергетической связи АТФ и других нуклеозидтрифосфатов (при этом возможно образование С-С-; C-S-; С-О-; и C-N- связей). В табл. 8.2 представлены шифры, принятые для различных ферментов, их систематические и тривиальные названия. В таблицу включены лишь ферменты, имеющие принципиальное значение при хранении, переработке сырья и в производстве пищевых продуктов. В дальнейшем, везде где это возможно, будут применяться тривиальные названия.
Внимание технологов, перерабатывающих биологическое сырье, привлекают прежде всего ферменты 1-го класса — оксидоредуктазы, а также 3-го класса — гидролазы, поскольку при переработке пищевого сырья происходит разрушение клеточной структуры биологического материала, повышается доступ кислорода воздуха к измельченным тканям и создаются Таблица. 8.2. Номенклатура ферментов, имеющих значение в пищевой промышленности ["Номенклатура ферментов", Рекомендации 1972 г. — М., 1979 (под ред. акад. А. Е Браунштейна)]
благоприятные условия для действия ферментов типа оксигеназ, а также высвобождаются гидролитические ферменты, которые активно расщепляют все основные структурные компоненты клетки (белки, липиды, полисахариды), в связи с чем процессы распада клеточного содержимого (процессы автолиза, самопереваривания) становятся преобладающими. Остановимся на рассмотрении отдельных представителей этих двух важнейших для пищевой промышленности классов ферментов с позиции описания их свойств, активности, механизма реакции и коснемся вопросов практического применения, которые будут рассмотрены более подробно в разделах, посвященных применению ферментов в конкретных пищевых технологиях. 295:: 296:: 297:: 298:: Содержание 298:: 299:: 300:: 301:: 302:: 303:: 304:: Содержание Оксидоредуктазы Полифенолоксидаза (Н.Ф. 1.14.18.1). Этот фермент известен под различными тривиальными названиями: о-дифенолоксидаза, тирозиназа, фенолаза, катехолаза и др. Фермент может катализировать окисление моно-, ди-, и полифенолов. Типичная реакция, катализируемая полифенолоксидазой, имеет вид:
Молекула фермента обладает четвертичной структурой и имеет молекулярную массу около 34 000 Да. Полифенолоксидаза — купропротеид. Содержание меди — 0,2%, или один атом Си на 1 молекулу фермента. Зона оптимальной активности лежит между рН 5,0 — 7,0. В зависимости от того, из какого источника выделен фермент, способность его к окислению различных фенолов различна. Более того, даже в одном и том же объекте Полифенолоксидаза может содержаться в виде различных молекулярных форм, отличающихся по способности к окислению различных фенолов. С действием этого фермента связано образование темноокрашенных соединений — меланинов при окислении кислородом воздуха аминокислоты — тирозина. Потемнение срезов картофеля, яблок, грибов, персиков и других растительных тканей в большей степени или полностью зависит от действия полифенолоксидазы. В пищевой промышленности основной интерес к этому ферменту сосредоточен на предотвращении рассмотренного нами ферментативного потемнения, которое имеет место при сушке плодов и овощей, а также при производстве макаронных изделий из муки с повышенной активностью полифенолоксидазы. Эта цель может быть достигнута путем тепловой инактивации фермента (бланшировка), добавлением ингибиторов (NaHSO3, SO2, NaCl) или связыванием субстрата посредством метилирования. Положительная роль фермента проявляется при некоторых ферментативных процессах: например, при ферментации чая. Окисление дубильных веществ чая под действием полифенолоксидазы приводит к образованию темноокрашенных и ароматических соединений, которые определяют цвет и аромат черного чая. Каталаза (Н.Ф. 1.11.1.6). Этот фермент катализирует разложение пероксида водорода в соответствии со следующей реакцией:
Таким образом, фермент окисляет одну молекулу перекиси водорода до кислорода с одновременным восстановлением другой молекулы перекиси водорода до Н2О. Каталаза относится к группе гемопротеиновых ферментов. Содержит 0,009% железа в виде геминовой группировки или 4 атома на одну молекулу фермента. Молекулярная масса ферментов, выделенных из различных объектов (дрожжей, растительных и животных тканей, микроорганизмов), лежит в пределах от 225 000 до 250 000 Да. Они имеют существенные различия в оптимуме рН (от 2 до 9), в термо- и рН-стабильности. Фермент ингибируется цианидом (обратимо), фенолами (обратимо лишь в слабой форме), щелочью и мочевиной (необратимо). Функцией каталазы в живом организме является защита клетки от губительного действия перекиси водорода. Хорошим источником для получения промышленных препаратов каталазы являются культуры микроорганизмов и печень крупного рогатого скота. Каталаза находит свое применение в пищевой промышленности при удалении избытка Н2О2 при обработке молока в сыроделии, где последняя используется в качестве консерванта; а также совместно с глюкозооксидазой применяется для удаления кислорода и следов глюкозы. Пероксидаза (Н.Ф. 1.11.1.7). Пероксидазы могут быть определены как ферменты, катализирующие следующую реакцию:
Пероксидаза — двухкомпонентный фермент, представляющий собой сочетание тема и гликопротеида. Показано, что углеводная часть придает белку большую специфичность; предполагают, что углеводы стабилизируют трехмерную структуру фермента. В настоящее время выделено и охарактеризовано большое число множественных форм фермента и доказано существование изоферментов, то есть тех форм ферментов, которые обусловлены генетически. В связи с этим принято говорить о целой системе пероксидаз, работающих в любом живом организме. Интересным представляется факт широкой субстратной специфичности пероксидаз по отношению к донорам водорода (1 -и субстрат), ими могут служить фенолы, амины, другие органические соединения; и строгой специфичности по отношению к акцептору водорода (2-й субстрат) — перекиси водорода. Механизм реакции, предположительно, основан на образовании комплексов фермент — донор и двух одновалентных ступеней окисления, как это отражает следующая схема: Пероксидаза + Н2О2 = Комплекс I Комплекс I + АН2 = Комплекс II + АН Комплекс II + АН = Пероксидаза + А Изучению пероксидазы были посвящены классические работы Г. Теореля, Б. Чанса, А. Н. Баха, Р. Шода. Наиболее активная Пероксидаза выделена из корней хрена. Ее молекулярная масса равна примерно 40 000 Да, изоэлектрическая точка 7,2. Фермент содержит один атом железа на молекулу. Он достаточно устойчив в растворах при величинах рН от 4 до 12; его термостабильность значительно выше термостабильности каталазы. Оптимум рН для пероксидазы хрена равен 7; при рН от 6 до 8 сохраняется 70% его активности. Липоксигеназа (Н.Ф.1.13.11.12). Этот фермент катализирует окисление полиненасыщенных высокомолекулярных жирных кислот (линолевой и линоленовой) кислородом воздуха с образованием высокотоксичных гидроперекисей. Ниже приведена реакция, катализируемая этим ферментом: R....... СН2-СН=СН-СН2-СН=СН-СН2 ....... СООН ↓ + О2 R....... СН2-СН=СН-СН=СН-С(ООН)Н-СН2 ....... СООН Возможно образование и циклических гидроперекисей по следующей схеме:
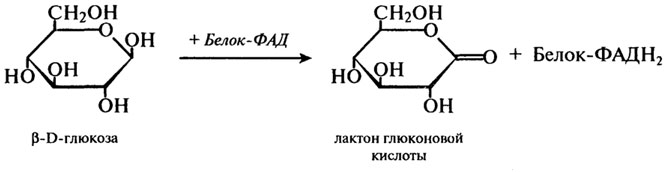
Однако основное количество жирных кислот превращаются в гидроперекиси, обладающие сильными окислительными свойствами, и именно на этом основано использование липоксигеназы в пищевой промышленности. Липоксигеназа впервые была выделена из семян сои в 1928 г. Последующие исследования показали, что липоксигеназа широко распространена и в других растительных объектах: пшенице и других злаках, в семенах масличных и бобовых культур, в картофеле, баклажанах и т. д. Тем не менее самым богатым источником фермента является мука соевых бобов. Липоксигеназа, полученная в кристаллическом состоянии из семян сои, имеет молекулярную массу 102 000 Да, изоэлектрическую точку 5,4. Оптимумы рН для ферментов, выделенных из различных объектов, сильно различаются. Оптимум температуры липоксигеназы находится между 20 и 30°С. В зерне пшеницы активность липоксигеназы колеблется в значительных пределах и является сортовым признаком. Кроме того, активность липоксигеназы связана с показателем жизнеспособности зерна. Она закономерно снижается со снижением всхожести зерна и может быть биохимическим тестом жизнеспособности семян. Значительная часть липоксигеназы пшеницы прочно связана с клейковинными белками и освобождается при обработке клейковинного комплекса раствором восстановленного глутатиона. Липоксигеназе принадлежит важная роль в процессах созревания пшеничной муки, связанных с улучшением ее хлебопекарных достоинств. Образующиеся под действием фермента продукты окисления жирных кислот способны вызывать сопряженное окисление ряда других компонентов муки (пигментов, SH-групп клейковинных белков, ферментов и др.). При этом происходит осветление муки, укрепление клейковины, снижение активности протеолитических ферментов и другие положительные изменения. В разных странах разработаны и запатентованы способы улучшения качества хлеба, основанные на использовании препаратов липоксигеназы (главным образом, липоксигеназы соевой муки). Все они требуют очень точного дозирования фермента, так как даже небольшая передозировка приводит к резко отрицательному эффекту и вместо улучшения качества хлеба происходит его ухудшение. Более мягкий способ воздействия на компоненты муки и теста связан с активацией собственной липоксигеназы муки путем некоторого варьирования технологического процесса. При этом исключается эффект передозировки фермента со всем комплексом нежелательных последствий. Использование липоксигеназы как улучшителя окислительного действия требует определенной осторожности, так как хорошо известна токсичность переокисленных жиров. Интенсивное окисление липоксигеназой свободных жирных кислот может сопровождаться вторичными процессами образования веществ различной химической природы с неприятным вкусом и запахом, характерным для прогорклого продукта. Технологически приемлема ограниченная степень окисления полиненасыщенных жирных кислот как промежуточного звена преобразования других компонентов биологического материала, не приводящая к накоплению фракции окисленных липидов. Глюкозооксидаза ( Н.Ф. 1.1.3.4). Этот фермент был впервые выделен еще в 1904 г. Н. А. Максимовым из плесневых грибов. Фермент представляет собой флавопротеид, в котором белок соединен с двумя молекулами ФАД. Он окисляет глюкозу с образованием в конечном счете глкжоновой кислоты и обладает практически абсолютной специфичностью по отношению к глюкозе. Суммарное уравнение имеет следующий вид: Глюкоза + Н2О + О2 = глюконовая кислота + Н2О2 Представленный выше процесс на самом деле протекает в несколько стадий: 1-я стадия:
2-я стадия:
3-я стадия: Белок-ФАД Н2 + О2 → Белок-ФАД + Н2О2 4-я стадия:
На первом этапе этой реакции происходит отнятие двух атомов водорода у первого углеродного атома глюкозы. При этом образуется восстановленный флавиновый фермент и лактон глюконовой кислоты. Далее восстановленный фермент реагирует с кислородом воздуха, и образуется перекись водорода. Токсичная перекись водорода расщепляется каталазой на кислород и воду, а β-О-глюконо-б-лактон подвергается спонтанному расщеплению с присоединением воды, в результате чего образуется глюконовая кислота. Высокоочищенные препараты глюкозооксидазы получают из плесневых грибов рода Aspergillus и Penicillium. Они имеют примерно одинаковую молекулярную массу — около 150 000 Да, изоэлектрическую точку 4,2 — 4,3 и оптимум рН 5,6. В последние годы глюкозооксидаза получила широкое применение. Благодаря исключительной специфичности препараты глюкозооксидазы применяются как аналитическое средство для количественного определения глюкозы. Кроме этого, препараты глюкозооксидазы нашли применение в пищевой промышленности как для удаления следов глюкозы, так и для удаления следов кислорода. Первое — необходимо при обработке пищевых продуктов, качество и аромат которых ухудшаются из-за того, что в них содержатся восстанавливающие сахара; например, при получении из яиц сухого яичного порошка. Здесь имеется в виду реакция Майяра, т. к. глюкоза при сушке и хранении яичного порошка, особенно при повышенной температуре, легко вступает в реакцию с аминными группами аминокислот и белков. Порошок темнеет, и образуется ряд веществ с неприятным вкусом и запахом. Второе — необходимо при обработке продуктов, в которых длительное присутствие небольших количеств кислорода приводит к изменению аромата и цвета (пиво, вино, фруктовые соки, майонез). Внесение пакетиков, содержащих смесь воды, глюкозы, фермента и буфера, способствует удалению кислорода из воздушного пространства. Во всех подобных случаях в ферментную систему включают каталазу, разлагающую Н2О2, которая образуется при реакции глюкозы с кислородом. Этот метод нашел широкое применение в США для удаления кислорода из банок с сухим молочным порошком. 298:: 299:: 300:: 301:: 302:: 303:: 304:: Содержание 303:: 304:: 305:: 306:: 307:: 308:: 309:: 310:: 311:: 312:: 313:: 314:: 315:: 316:: 317:: 318:: 319:: 320:: 321:: 322:: 323:: Содержание Гидролитические ферменты Роль ферментов класса гидролаз в пищевых технологиях очень велика. Это находит отражение в специальной литературе, монографиях, технических инструкциях, стандартах. Поэтому в этом разделе остановимся на краткой характеристике наиболее важных представителей гидролитических ферментов. Для технологов наибольший интерес представляют три подкласса ферментов класса гидролаз. Это ферменты, действующие на сложноэфирные связи — эстеразы (Н.Ф.3.1); действующие на гликозидные соединения — гликозидазы (Н.Ф.3.2) и действующие на пептидные связи — протеазы (Н.Ф.3.4). Эстеразы (Н.Ф.3.1). Этот подкласс включает большое число ферментов (около 150), которые разделены на семь подподклассов: ферменты, действующие на эфиры карбоновых кислот (3.1.1); эстеразы тиоловых эфиров (3.1.2); гидролазы фосфорных моноэфиров или фосфатазы (3.1.3); гидролазы фосфорных диэфиров (3.1.4); гидролазы моноэфиров олигофосфорных кислот (3.1.5); сульфатазы (3.1.6); эстеразы моноэфиров дифосфорных кислот (3.1.7). Наиболее важными с точки зрения участия в различных биохимических процессах, имеющих место при хранении и переработке пищевого сырья, являются ферменты подподкласса 3.1.1. Липаза (Н.Ф.3.1.1.3). Липаза или триацилглицероллипаза широко распространена в природе и играет важную роль в процессах, протекающих при переработке и хранении пищевых продуктов. В настоящее время выделены и охарактеризованы липазы растительного происхождения (липаза клещевины, пшеницы и других злаков), животного (панкреатическая липаза, липаза молока) и микробного (бактериальные и грибные липазы). Обычно липазы катализируют реакцию расщепления триглицеридов согласно приведенному ниже суммарному уравнению:
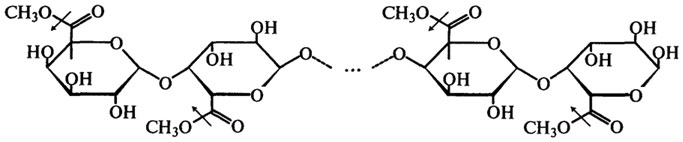
Причем предпочтительнее гидролизуются связи в положении 3 и 1 и лишь затем в положении 2. Многочисленные экспериментальные данные дают основание предположить следующий путь липолиза: триглицерид → 1,2-диглицерид → 2-моноглицерид → глицерин Установлено, что липазы быстрее отщепляют остатки высокомолекулярных жирных кислот, чем низшие карбоновые кислоты. Ферментативный гидролиз липидов имеет существенное отличие от других гидролитических реакций. Парадокс заключается в том, что липаза — водорастворимый фермент, а ее субстрат гидрофобен; однако активность липазы возрастает на границе "вода — липид". Этот феномен известен под названием "межфазная активация". Липазы различного происхождения сильно отличаются друг от друга по специфичности действия, сродству к различным субстратам, растворимости, оптимуму рН и другим свойствам. Так, например, липаза семян клещевины нерастворима в воде, имеет оптимум рН 4,7 — 5,0; панкреатическая липаза растворима, и оптимум рН ее действия лежит в слабощелочной среде. Липазы микробного происхождения и липаза пшеничных зародышей также отличаются от липазы клещевины. Они растворимы в воде и имеют рН оптимум при 8,0. Липаза молока, молекулярная масса которой примерно 7000 Да, имеет оптимум рН 9,0 — 9,2 при гидролизе молочного жира. Зерновая липаза участвует в процессе порчи зерновых продуктов при хранении. Особенно это касается продуктов, содержащих повышенное количество жира, например, овсяной муки или крупы, пшена. Накопление свободных жирных кислот под действием липазы (рост кислотного числа жира) — признак ухудшения качества продукта. Свободные жирные кислоты, особенно ненасыщенные, легко подвергаются окислению под воздействием разных факторов: липоксигеназы, тепловой обработки, кислорода воздуха, солнечного света и др. Таким образом, липазы могут инициировать процесс прогоркания и ограничивать сроки хранения пищевых продуктов. Одна из особенностей липаз связана с тем, что эти ферменты способны катализировать и обратную реакцию, осуществлять синтез сложных эфиров, а также производить переэтерефикацию триглицеридов, т. е. изменять их жирнокислотный состав. На этом основании разрабатываются способы получения новых форм жировых продуктов с использованием специфических липаз. Так, например, путем реакции переэтерифи-кации делаются попытки получения жира — аналога масла какао из дешевого исходного сырья. Пектинэстераза (Н.Ф.3.1.1.11). Пектинэстеразы синтезируются высшими растениями, микроскопическими грибами, дрожжами и бактериями. Пектинэстераза катализирует гидролиз сложноэфирных связей в молекуле растворимого пектина, в результате чего образуется метиловый спирт и полигалактуроновая кислота. Процесс протекает согласно следующей схеме (стрелками показано действие фермента):
Таким образом, пектинэстераза отщепляет метоксильные группы от метоксилированной полигалактуроновой кислоты (см. также гл. Углеводы). Желирующая способность пектина зависит от степени метоксилирования или степени этерификации, поэтому действие пектинэстеразы по отщеплению метоксильных групп приводит к снижению желирующей способности и сопровождается падением вязкости. На этом, очевидно, и основывается применение этого фермента для осветления плодовых соков и вина. Обычно комплексные препараты пектолитических ферментов, применяемые для этих целей, получают из различных плесневых грибов, и прежде всего из A. niger. Гидролазы гликозидов или гликозидазы (Н.Ф.3.2). Этот подкласс включает около ста ферментов с разной специфичностью действия, осуществяющих гидролиз олиго- и полисахаридов; некоторые ферменты этого типа способны осуществлять трансферазные реакции — переносить гликозидные остатки на олиго- и полисахариды, наращивать полисахаридные цепочки. Представители гликозидаз были одними из первых ферментов, обратимость действия которых in vitro была экспериментально доказана. Основной формой запасных углеводов в семенах и клубнях растений является крахмал. Ферментативные превращения крахмала лежат в основе многих пищевых технологий. Поэтому ферменты амилолитического комплекса растительного, животного и микробного происхождения интенсивно изучаются со времени их открытия Кирхгофом в 1814 г.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-04-26; просмотров: 953; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.31.48 (0.022 с.) |