Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Теория химического строения А.М. Бутлерова. Электронное строение атома углерода и виды гибридизации.Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Теория химического строения А.М. Бутлерова. Электронное строение атома углерода и виды гибридизации. В 1861 году А.М. Бутлеровым была предложена теория химического строения органических соединений, которая состоит из следующих основных положений. 1. В молекулах веществ существует строгая последовательность химического связывания атомов, которая называется химическим строением. 2. Химические свойства вещества определяются природой элементарных составных частей, их количеством и химическим строением. 3. Если у веществ с одинаковым составом и молекулярной массой различное строение, то возникает явление изомерии. 4. Так как в конкретных реакциях изменяются только некоторые части молекулы, то исследование строения продукта помогает определить строение исходной молекулы. 5. Химическая природа (реакционная способность) отдельных атомов в молекуле меняется в зависимости от окружения, т.е. от того, с какими атомами других элементов они соединены.
Атом углерода в возбужденном состоянии содержит четыре неспаренных электрона на внешнем энергетическом уровне и способен образовать четыре ковалентных связи.
В образовании связей участвуют гибридные орбитали. Первое валентное состояние – sp3-гибридизация. В результате гибридизации с участием одной s и трех p орбиталей атома углерода образуются четыре эквивалентные sp3-гибридные орбитали, направленные к вершинам тетраэдра под углами 109,5о: В состоянии sp3-гибридизации атом углерода образует четыре s -связи с четырьмя заместителями и имеет тетраэдричекую конфигурацию с валентными углами, равными или близкими 109,5о. (например, метан) Второе валентное состояние – sp2-гибридизация. В результате гибридизации с участием одной s- и двух p-орбиталей атома углерода образуются три эквивалентные sp2-гибридные орбитали, лежащие в одной плоскости под углами 120о, а не участвующая в гибридизации p-орбиталь расположена перпендикулярно плоскости гибридных орбиталей. В состоянии sp2-гибридизации атом углерода образует три s -связи за счет гибридных орбиталей и одну p -связь за счет не участвующей в гибридизации p-орбитали и имеет три заместителя. (например, этилен) Третье валентное состояние углерода – sp-гибридизация. В результате гибридизации с участием одной s- и одной p–орбитали образуются две эквивалентные sp-гибридные орбитали, лежащие под углом 1800, а не участвующие в гибридизации p-орбитали расположены перпендикулярно плоскости гибридных орбиталей и друг другу. В состоянии sp-гибридизации атом углерода образует две s -связи за счет гибридных орбиталей и две p -связи за счет не участвующих в гибридизации p-орбиталей и имеет два заместителя. (например, ацетилен)
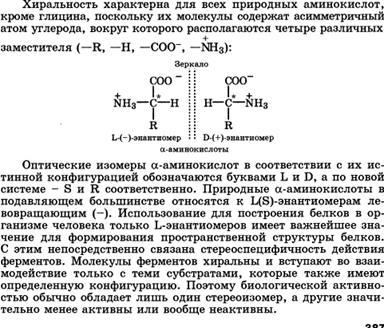
Понятие о конфигурации молекул. Оптическая, или зеркальная изомерия. Элементы симметрии молекул (ось, плоскость, центр). Ассиметрический атом углерода как центр хиральности. Оптическая активность и удельное вращение веществ.
Молекулы с одним центром хиральности (энантиомерия). Глицериновый альдегид как кон-фигурационный стандарт. Проекционные формулы Фишера. Относительная и абсолютная конфигурация. D-, L- и R-, S-системы. Понятие о рацематах. Энантиомеры - стереоизомеры, относящиеся друг к другу, как предмет и несовместимое с ним зеркальное отображение.
В виде энантиомеров могут существовать только хиральные молекулы.
Хиральность - это свойство объекта быть несовместимым со своим зеркальным отражением. Хиральными (от греч. cheir - рука), или асимметричными, объектами являются левая и правая рука, а также перчатки, ботинки и др. Эти парные предметы представляют собой объект и его зеркальное отражение (рис. 7.1, а). Такие предметы не могут быть полностью совмещены друг с другом.
В то же время существует множество окружающих нас предметов, которые совместимы со своим зеркальным отражением, т. е. они являются ахиральными (симметричными), например тарелки, ложки, стаканы и т. д. Ахиральные предметы обладают по крайней мере одной плоскостью симметрии, которая делит объект на две зеркальноидентичные части (см. рис. 7.1, б).
Подобные взаимоотношения наблюдаются также в мире молекул, т. е. молекулы делятся на хиральные и ахиральные. У ахиральных молекул есть плоскости симметрии, у хиральных их нет.
В хиральных молекулах имеется один или несколько центров хиральности. В органических соединениях в качестве центра хиральности чаще всего выступает асимметрический атом углерода.
Асимметрическим является атом углерода, связанный с четырьмя различными атомами или группами.
При изображении стереохимической формулы молекулы символ «С» асимметрического атома углерода обычно опускается.
Для изображения конфигурационных изомеров на плоскости можно пользоваться стереохимическими формулами. Однако удобнее применять более простые в написании проекционные формулы Фишера (проще - проекции Фишера). Тетраэдрическую модель одного из энантиомеров располагают в пространстве так, чтобы цепь атомов углерода оказалась в вертикальном положении, а карбоксильная группа - сверху. Связи с неуглеродными заместителями (Н и ОН) у хирального центра должны быть направлены к наблюдателю. После этого модель проецируют на плоскость. Символ асимметрического атома при этом опускается, под ним понимают точку пересечения вертикальной и горизонтальной линий. Тетраэдрическую модель хиральной молекулы перед проецированием можно располагать в пространстве по-разному,. Необходимо только, чтобы связи, образующие на проекции горизонтальную линию, были направлены к наблюдателю, а вертикальные связи - за плоскость рисунка. • в проекционной формуле разрешается менять местами два любых заместителя у одного и того же хирального центра четное число раз (двух перестановок бывает достаточно); • проекционную формулу разрешается поворачивать в плоскости рисунка на 180? (что эквивалентно двум перестановкам), но не на 90?. За конфигурационный стандарт был принят глицериновый альдегид. Его левовращающему энантиомеру была произвольно приписана формула (I). Такая конфигурация атома углерода была обозначена буквой l (от лат. laevus - левый). Правовращающему энантиомеру соответственно была приписана формула (II), а конфигурация обозначена буквой d (от лат. dexter - правый). Заметим, что в стандартной проекционной формуле l-глицеринового альдегида группа ОН находится слева, а у d-глицеринового альдегида - справа.
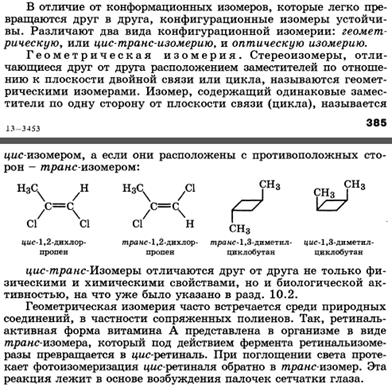
Отнесение к d- или l-ряду других родственных по структуре оптически активных соединений производится путем сравнения конфигурации их асимметрического атома с конфигурацией d- или l-глицеринового альдегида. Например, у одного из энантиомеров молочной кислоты (I) в проекционной формуле группа ОН находится слева, как у l-глицеринового альдегида, поэтому энантиомер (I) относят к l-ряду. Из тех же соображений энантиомер (II) относят к d-ряду. Так из срав- нения проекций Фишера определяют относительную конфигурацию.
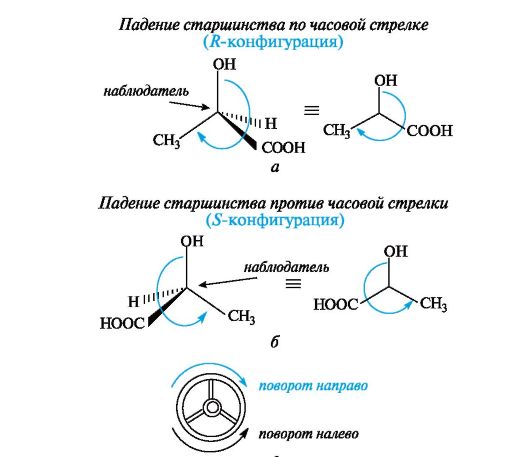
Следует отметить, что l-глицериновый альдегид имеет левое вращение, а l-молочная кислота - правое (и это не единичный случай). Более того, одно и то же вещество может быть как лево-, так и правовращающим в зависимости от условий определения (разные растворители, температура). Знак вращения плоскости поляризованного света не связан с принадлежностью к d- или l-стереохимическому ряду. Практическое определение относительной конфигурации оптически активных соединений проводят с помощью химических реакций: либо исследуемое вещество превращают в глицериновый альдегид (или другое вещество с известной относительной конфигурацией), либо, наоборот, изd- или l-глицеринового альдегида получают исследуемое вещество. Разумеется, что в ходе всех этих реакций не должна изменяться конфигурация асимметрического атома углерода. Произвольное приписание лево- и правовращающему глицериновому альдегиду условных конфигураций было вынужденным шагом. В то время абсолютная конфигурация не была известна ни для одного хирального соединения. Установление абсолютной конфигурации стало возможным только благодаря развитию физико-химических методов, особенно рентгеноструктурного анализа, с помощью которого в 1951 г. впервые была определена абсолютная конфигура,ция хиральной молекулы - это была соль (+)-винной кислоты. После этого стало ясно, что абсолютная конфигурация d- и l-глицериновых альдегидов действительно такая, какая им была первоначально приписана. d,l-Система в настоящее время применяется для α-аминокислот, гидроксикислот и (с некоторыми дополнениями) для углеводов R,S-Система обозначения конфигурации. d,L-Система имеет весьма ограниченное применение, так как часто невозможно соотнести конфигурацию какого-либо соединения с глицериновым альдегидом. Универсальной системой обозначения конфигурации центров хиральности является R,S-система (от лат. rectus - прямой, sinister - левый). В ее основе лежит правило последовательности, основанное на старшинстве заместителей, связанных с центром хиральности. Старшинство заместителей определяется атомным номером элемента, непосредственно связанного с центром хиральности, - чем он больше, тем старше заместитель. Так, группа ОН старше NH2, которая, в свою очередь, старше любой алкильной группы и даже СООН, поскольку в последней с асимметрическим центром связан атом углерода. Если атомные номера оказываются одинаковыми, старшей считается группа, у которой следующий за углеродом атом имеет больший порядковый номер, причем, если этот атом (обычно кислород) связан двойной связью, он учитывается дважды. В результате следующие группы так располагаются в порядке падения старшинства: -СООН > -СН=О > -СН2ОН. Для определения конфигурации тетраэдрическую модель соединения располагают в пространстве так, чтобы самый младший замес- титель (в большинстве случаев это атом водорода) был наиболее удален от наблюдателя. Если старшинство трех остальных заместителей убывает по часовой стрелке, то центру хиральности приписывают R-конфигурацию (рис. 7.4, а), если против часовой стрелки - S -конфигурацию (см. рис. 7.4, б), как это видно водителю, находящемуся за рулем (см. рис. 7.4, в).
Рис. 7.4. Определение конфигурации энантиомеров молочной кислоты по R,S- системе (объяснение в тексте) Для обозначения конфигурации по RS-системе можно применить проекции Фишера. Для этого проекцию преобразуют так, чтобы
Вопрос 9 Кислотность и основность органических соединений. Для оценки кислотности и основности органических соединений наибольшее значение имеют две теории – теория Бренстеда и теория Льюиса. По теории Льюиса кислотные и основные свойства соединений определяются их способностью принимать или отдавать пару электронов с образованием связи. В соответствии с принципом ЖМКО кислоты и основания Льюиса делятся на жесткие и мягкие. Кислотами Льюиса могут быть атомы, молекулы или катионы, обладающие вакантной орбиталью и способные принимать пару электронов с образованием ковалентной связи. Кислоты Льюиса – акцепторы пары электронов; основания Льюиса – доноры пары электронов. Основания Льюиса (атом, молекула или анион) должны обладать по крайней мере одной парой валентных электронов, которую они способны предоставить партнеру для образования ковалентной связи. Все основания Льюиса представляют собой нуклеофильные реагенты. По теории Бренстеда (протолитической теории) кислотность и основность соединений связывается с переносом протона Н+. Кислота и основание образуют сопряженную кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряженное ей основание, и напротив, чем сильнее основание, тем слабее сопряженная ему кислота. Кислоты Бренстеда (протонные кислоты) – нейтральные молекулы или ионы, способные отдавать протон (доноры протонов). Основания Бренстеда – нейтральные молекулы или ионы, способные присоединять протон (акцепторы протонов). Кислотность и основность являются не абсолютными, а относительными свойствами соединений: кислотные свойства обнаруживаются лишь в присутствии основания; основные свойства – только в присутствии кислоты. В качестве растворителя при изучении кислотно-основных равновесий обычно используется вода. В зависимости от природы элемента, с которым связан протон, различают ОН- кислоты (карбоновые кислоты, фенолы, спирты), SH-кислоты (тиолы), NН-кислоты (амины, амиды, имиды), СН-кислоты (углеводороды и их производные). Элемент и связанный с ним атом водорода называют кислотным центром. Во всех случаях присутствует сдвиг электронной плотности от атома водорода к более электроотрицательному атому, протону более или менее легко отщепиться. Чем выше электроотрицательность элемента, с которым связан протон, тем больше кислотность соединения (так, карбоновые кислоты являются более сильными кислотами, чем тиолы или амины). Наличие в молекуле электроноакцепторных групп, обладающих отрицательными электронными эффектами, увеличивает положительный заряд на протоне, что приводит к усилению кислотных свойств. Для образования ковалентной связи с протоном основания Бренстеда должны предоставлять либо неподелениую пару электронов, либо электроны p-связи. В соответствии с этим основания Бренстеда делятся на п -основания и p-основания. n -основания могут быть нейтральными или отрицательно заряженными. Как правило, анионы обладают более сильно выраженным основным характером, чем нейтральные вещества. То есть амид-ион NН2– или гидроксид-ион НО– по основности превосходят аммиак NН3 и воду Н2О. В p-основаниях, к которым относятся алкены, алкадиены, арены, центром основности, т.е. местом присоединения протона, являются электроны p-связи. Это очень слабые основания, так как протонируемые электронные пары несвободны. Наличие электронодонорных заместителей увеличивает основность органических соединений. 1. Зависимость кислотности от гетероатома. Под природой гетероатома понимают его электроотрицательность (Э.О.) и поляризуемость. Чем больше (Э.О.) тем легче осуществляется гетеролитический разрыв в молекуле. В периодах слева направо с ростом заряда ядра растет (Э.О), т.е. способность элементов удерживать отрицательный заряд. В результате смещения электронной плотности связь между атомами поляризуется. Чем больше электронов и чем больше радиус атома, тем дальше электроны внешнего энергетического уровня расположены от ядра, тем выше поляризуемость и выше кислотность. Пример: СН- NH- OH- SH-
увеличение Э.О. и кислотности С, N,О – элементы одного периода. Э.О. по периоду растет, кислотность увеличивается. В этом случае поляризуемость влиять на кислотность не будет. Поляризуемость атомов в периоде изменяется незначительно, поэтому главным фактором определяющим кислотность является Э.О. Теперь рассмотрим ОН- SH-
увел-е кислотности О, S – находятся в одной группе, радиус в группе сверху вниз увеличивается, следовательно, растет и поляризуемость атома, что ведет к увеличению кислотности. У S радиус атома больше, чем у О, поэтому тиолы проявляют более сильные кислотные свойства по сравнению со спиртами. 2. Влияние углеводородного радикала и присутствующих в нем заместителей Электроноакцепторные (Э.А.) заместители способствуют делокализации электронной плотности, что ведёт к стабильности аниона и соответственно увеличению кислотности. Электронодонорные (Э.Д.) заместители наоборот способствуют концентрации электронной плотности в кислотном центре, что ведет к понижению кислотности и увеличению основности. Влияние растворителя. Взаимодействие молекул или ионов растворенного вещества с растворителем называется процессом сольватации. Стабильность аниона существенно зависит от его сольватации в растворе: чем больше ион сольватирован, тем он устойчивее, а сольватация тем больше, чем меньше размер иона и чем меньше делокализация в нем отрицательного заряда.
Кислотные свойства 1. С активными металлами: HO-CH2-CH2-OH + 2Na → H2↑+ NaO-CH2-CH2-ONa (гликолят натрия) 2. С гидроксидом меди(II) – качественная реакция!
Качественной реакцией на двухатомные и многоатомные спирты (диольный фрагмент) является реакция с Си(ОН)2 в щелочной среде, в результате которой образуется комплексное соединение гликолят меди в растворе, дающем синее окрашивание. Упрощённая схема
Основные свойства 1. С галогенводородными кислотами HO-CH2-CH2-OH + 2HCl H+ ↔ Cl-CH2-CH2-Cl + 2H2O С азотной кислотой
Тринитроглицерин - основа динамита Этиленгликольтоксичен – сильный Яд! Угнетает ЦНС и поражает почки. Глицерин (пропантриол-1,2,3) – не ядовит. Без запаха. Хорошо смешивается с водой. Распространён в живой природе. Играет важную роль в обменных процессах, так как входит в состав жиров (липидов) животных и растительных тканей. Применяется как компонент мазей для смягчения кожи. Многоатомный циклический спирт инозит относится к витаминоподобным соединениям (витамины группы В) и является структурным компонентом сложных липидов – фосфатидилинозитов. Этилендиамин применяется для получения этилендиаминтетрауксусной кислоты взаимодействием с хлоруксусной кислотой. Его соли с жирными кислотами используются как смягчающие агенты при производстве текстиля. Также этилендиамин применяется в производстве красителей, эмульгаторов, стабилизаторов латексов, пластификаторов и фунгицидов. Этилендиамин токсичен; предельно допустимая концентрация его паров в воздухе составляет 0,001 мг/л. Из полиаминов аиболее известны тетраметилендиамин, или путресцин H2N(CH2)4NH2, и пентаметилендиамин, или кадаверин H2N(CH2)5NH2. Их долгое время считали трупными ядами, т.е. веществами, образующимися при декарбоксилировании диаминокислот и обусловливающими ядовитость гниющих белков.
12. Фенолы.Общая характеристика. Электронное строение карбонильной группы. Реакции нуклеофильного присоединения АN по карбонильной группе. Реакции присоединения воды, синильной кислоты, спиртов, би-сульфита натрия. Механизм альдольной конденсации и реакции Канницаро.
Строение карбонильной группы C=O. · Свойства альдегидов и кетонов определяются строением карбонильной группы >C=O.
Связь С=О сильно полярна. Ее дипольный момент значительно выше, чем у связи С–О в спиртах. Электроны кратной связи С=О, в особенности более подвижные p-электроны, смещены к электроотрицательному атому кислорода, что приводит к появлению на нем частичного отрицательного заряда. Карбонильный углерод приобретает частичный положительный заряд.
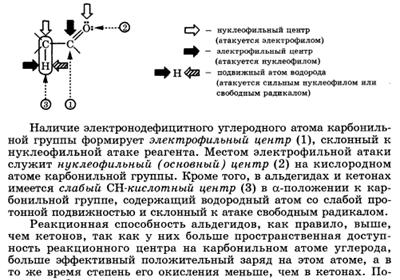
· Поэтому углерод подвергается атаке нуклеофильными реагентами, а кислород - электрофильными, в том числе Н+. В молекулах альдегидов и кетонов отсутствуют атомы водорода, способные к образованию водородных связей. Поэтому их температуры кипения ниже, чем у соответствующих спиртов. Метаналь (формальдегид) - газ, альдегиды С2–C5 и кетоны С3–С4 - жидкости, высшие - твердые вещества. Низшие гомологи растворимы в воде, благодаря образованию водородных связей между атомами водорода молекул воды и карбонильными атомами кислорода. С увеличением углеводородного радикала растворимость в воде падает.
Нуклеофильное присоединение Для альдегидов и кетонов наиболее характерны реакции нуклеофильного присоединения AN. Общее описание механизма нуклеофильного присоединения AN Легкость нуклеофильной атаки по атому углерода карбонильной группы альдегида или кетона зависит от величины частичного положительного заряда на атоме углерода, его пространственной доступности и кислотно-основных свойств среды.
С учетом электронных эффектов групп, связанных с карбонильным атомом углерода, величина частичного положительного заряда δ+ на нем в альдегидах и кетонах убывает в следующем ряду:
Пространственная доступность карбонильного атома углерода уменьшается при замене водорода более объемистыми органиче- скими радикалами, поэтому альдегиды более реакционноспособны, чем кетоны.
Общая схема реакций нуклеофильного присоединения AN к карбонильной группе включает нуклеофильную атаку по карбонильному атому углерода, за которой следует присоединение электрофила к атому кислорода.
В кислой среде активность карбонильной группы, как правило, увеличивается, поскольку вследствие протонирования атома кислорода на атоме углерода возникает положительный заряд. Кислотный катализ используют обычно тогда, когда атакующий нуклеофил обладает низкой активностью.
По приведенному выше механизму осуществляется ряд важных реакций альдегидов и кетонов.
Присоединение спиртов. Спирты при взаимодействии с альдегидами легко образуют полуацетали. Полуацетали обычно не выделяют из-за их неустойчивости. При избытке спирта в кислой среде полуацетали превращаются в ацетали.
Применение кислотного катализатора при превращении полуацеталя в ацеталь становится понятным из приведенного ниже механизма реакции. Центральное место в нем занимает образование карбо- катиона (I), стабилизированного за счет участия неподеленной пары электронов соседнего атома кислорода (+M-эффект группы С2Н5О).
Реакции образования полуацеталей и ацеталей обратимы, поэтому ацетали и полуацетали легко гидролизуются избытком воды в кислой среде. В щелочной среде полуацетали устойчивы, так как алкоксидион является более трудно уходящей группой, чем гидроксид-ион. Присоединение воды. Присоединение воды к карбонильной группе - гидратация - обратимая реакция. Степень гидратации альдегида или кетона в водном растворе зависит от строения субстрата.
Трихлороуксусный альдегид (хлораль) гидратирован полностью. Электроноакцепторная трихлорометильная группа настолько стабилизирует хлоральгидрат, что это кристаллическое вещество отщепляет воду только при перегонке в присутствии дегидратирующих веществ - серной кислоты и др.
Присоединение аминов и их производных. Амины и другие азотсодержащие соединения общей формулы NH2X (X = R, NHR) реагируют с альдегидами и кетонами в две стадии. Сначала образуются продукты нуклеофильного присоединения, которые затем вследствие неустойчивости отщепляют воду. В связи с этим данный процесс в целом классифицируют как реакцию присоединения-отщепления.
В случае первичных аминов получаются замещенные имины (их называют также основаниями Шиффа). Имины - промежуточные продукты многих ферментативных процессов. Получение иминов проходит через стадию образования аминоспиртов, которые бывают относительно устойчивы, например при взаимодействии формальдегида с α-аминокислотами (см. 12.1.4).
Имины являются промежуточными продуктами получения аминов из альдегидов и кетонов путем восстановительного аминирования. Этот общий способ заключается в восстановлении смеси карбонильного соединения с аммиаком (или амином). Процесс протекает по схеме присоединения-отщепления с образованием имина, который затем восстанавливается в амин.
При взаимодействии альдегидов и кетонов с производными гидразина получаются гидразоны. Эту реакцию можно использовать для выделения альдегидов и кетонов из смесей и их хроматографической идентификации.
Образование бисульфитных соединений Присоединением молекулы кислого сернистокислого натрия(бисульфита) получаются так называемые бисульфитные соединения, причем водород присоединяется ккислороду карбонильной группы, а остаток SO2ONa — к углеродному атому:
В бисульфитных соединениях атом серы непосредственно связан с углеродом.
КАННИЦЦАРО РЕАКЦИЯ, окислит.-восстановит. диспропорционирование альдегидов под действием щелочис образованием первичных спиртов и карбоновых к-т, напр.:
Дезаминирование, Внутримолекулярное дезаминирование R-CH2 – CH(NH2) - COOH→ R- CH=CH-COOH + NH3 α,β – ненасыщенная кислота Гидролитическое дезаминирование R-CH(NH2) – COOH +H2O → R – CH(OH) – COOH + NH3 α - оксикислота Окислительное дезаминирование R-CH(NH2) – COOH +1/2 O2→ R –C(O) – COOH + NH3 α-кетокислота
Образование комплексов с металлами. α-Аминокислоты образуют с катионами тяжелых металлов внутрикомплексные соли. Со свежеприготовленным гидроксидом меди(II) все α-аминокислоты в мягких условиях дают хорошо кристаллизующиеся внутрикомплексные (хелатные) соли меди(II) синего цвета:
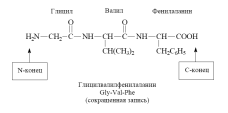
В таких солях ион меди координационными связями соединен с аминогруппами. Образование пептидной связи. Межмолекулярное взаимодействие -аминокислот приводит к образованию пептидов. При взаимодействии двух -аминокислот образуется дипептид.
Межмолекулярное взаимодействие трех -аминокислот приводит к образованию трипептида и т.д.
Фрагменты молекул аминокислот, образующие пептидную цепь, называются аминокислотными остатками, а связь CO–NH - пептидной связью. 22. Декарбоксилирование α-аминокислот – образование биогенных аминов и биорегуляторов (гиста-мин, триптамин).
Реакции декарбоксилирования необратимы и катализируются ферментами декарбоксилазами. Простетическая группа декарбоксилаз в клетках животных - пиридоксальфосфат. Амины, образовавшиеся при декарбоксилировании аминокислот, часто являются биологически активными веществами. Они выполняют функцию нейромедиаторов (серотонин, дофамин, ГАМК и др.), гормонов (норадреналин, адреналин), регуляторных факторов местного действия (гистамин, карнозин, спермин и др.). Гистамин образуется при декарбоксилировании аминокислоты гистидина. Он синтезируется в тучных клетках, накапливается в секреторных гранулах, выделяется при раздражении клеток.
Гистамин оказывает разнообразные биологические эффекты: вызывает расширение сосудов, снижает артериальное давление, увеличивает тканевую проницаемость, вызывает местный отёк, стимулирует желудочную секрецию, обладает бронхоспатическим эффектом. В высокой концентрации он является медиатором воспалительных и аллергических реакций. Серотонин образуется при декарбоксилировании гидрокситриптофана. Он синтезируется в хромаффиннных клетках, в некоторых ядрах подкорковых структур, тромбоцитах.
Эффекты серотонина: вызывает спазм сосудов, повышение артериального давления, стимулирует перистальтику кишечника, участвует в терморегуляции, в механизмах сна, является источником для синтеза гормона мелатонина, влияет на психические реакции человека. Так, при шизофрении наблюдается нарушение обмена серотонина. Катехоламины (дофамин, адреналин, норадреналин) синтезируются из аминокислоты тирозина.
Дофамин – возбуждающий медиатор, при его дефиците развивается болезнь Паркинсона (адинамия, ригидность, тремор). Адреналин вызывает спазм сосудов, повышают артериальное давление, стимулирует работу сердца, является гормоном. Норадреналин в основном выполняет нейромедиаторные функции. Полиамины (спермин, спермидин) синтезируются из орнитина и метионина, входят в состав хроматина, участвует в регуляции процессов трансляции, транскрипции, репликации. Так как биогенные амины очень активны, они быстро инактивируются в тканях. Распад биогенных аминов осуществляется несколькими способами: окисление, метилирование, дезаминирование. Основным способом инактивации биогенных аминов является окислительное дезаминирование под действием ферментов аминооксидаз (моноаминооксидаз, полиаминооксидаз).
Аминокислоты могут ковалентно связываться друг с другом с помощьюпептидных связей. Карбоксильная группа одной аминокислоты ковалентно связывается с аминогруппой другой аминокислоты. При этом возникает R-CO-NH-R связь, называемая пептидной связью. При этом происходит отщепление молекулы воды.
O- и N-глюкозиды. Гидролиз глюкозидов. Фосфаты моносахаридов. Ацилирование аминосаха-ров. Окисление моносахаридов. Получение озазонов глюкозы. Восстановительные свойства аль-доз. Ксилит, сорбит. Аскорбиновая кислота. Гликозиды – производные циклич.форм углеводов, в которых полуацетальная гидроксильная группа заменена группой ОR. Неуглевод.комонент – агликон. Связь между аномерным центром и группой –ОR – гликозидная. Подразделяют на пиранозиды фуранозиды. Гликозиды глюкозы называют глюкозидами, рибозы – рибозидами и т.д.
с а х а р агликон (чаще моносахарид) (спирт, ароматич.соед., стероиды и т.д.)
Гликозид синигрин; гидролиз:
Фосфаты моносахарид. Большое значение имеют эфиры фосфорной кислоты – фосфаты. Они содержатся во всех растительных и животных организмах и представляют собой метаболически активные формы моносахаридов. Наиболее важную роль играют фосфаты D-глюкозы и D-фруктозы.
Окисление глюкозы внейтральной, слабо-кислой среде:
Окисление с помощью сильного окислителя:
Окисление глюкозы в щелочной среде. Р-ция Толленса:
Реакция Фелинга:
Получение озазона глюкозы (р-ция с фенилгидразином):
Восстановление. При восстановлении моносахаридов образуются альдиты.
Альдиты легко растворимы в воде, обладают сладким вкусом, некоторые из них (ксилит и сорбит) используются как заменители сахара для больных сахарным диабетом. При восстановлении альдоз получается лишь один полиол. Ксилит и сорбит – многоатомные спирты. Заменители сахара для больных диабетом.
Аскорбиновая кислота(витамин С).
По структуре близок к моносахаридам. Представляет собой γ-лактон кислоты. Содержится во фруктах, особенно в цитрусовых, ягодах(шиповникэ, черная смородина), овощах, молоке. Проявляет сильные кислотные свойства за счет одной из гидроксильных групп ендиольного фрагмента. При образовании солей γ-лактонное кольцо не размыкается. Обладает сильными восстановительными свойствами. Образующаяся при ее окислении дегидроаскорбиновая кислота легко восстанавливается в аскорбиновую. Этот процесс обеспечивает ряд окислительно-восстановительных реакции в организме.
Гетероциклы с одним гетероатомом. Пиррол, индол, пиридин, холин. Понятие о строении тетрапиррольных соединений (порфин, гем). Производные пиридина (никотинами
|
||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-01; просмотров: 1397; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.214 (0.014 с.) |






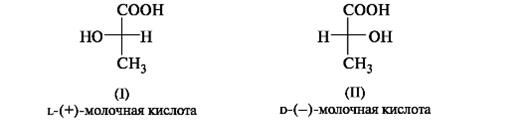










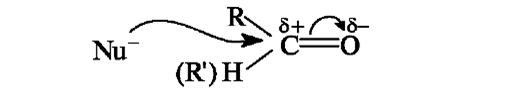




























 ГЛИКОЗИДЫ
ГЛИКОЗИДЫ Гликозид ванилина; гидролиз:
Гликозид ванилина; гидролиз: