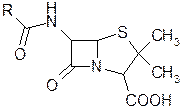Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Современные принципы разработки лекарственных препаратов или драг-дизайнСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Введение Предмет медицинской химии
Медицинская химия – одна из областей органической химии, связанная с проблемой конструирования будущих лекарственных препаратов. Она зародилась в конце XIX – начале XX века и по-настоящему сформировалась к 70-м годам прошлого столетия, когда возникла соответствующая система понятий и определений. Предметом медицинской органической химии является синтез и идентификация потенциально биологически активных веществ, выявление взаимосвязи между их химической структурой и физиологической активностью, а также решение обратной задачи: конструирование необходимых структур, обладающих определёнными физико-химическими свойствами, оптимальными фармакокинетическими параметрами и заданной биологической активностью. Основной акцент делается на лекарства, но интересы медицинской химии не ограничиваются лекарствами, а включают биологически активные соединения вообще. Предметом медицинской химии является также изучение, идентификация и синтез продуктов метаболизма этих лекарств и родственных соединений. Медицинская химия - наука междисциплинарная и находится на границе органической химии с такими уже сформировавшимися и признанными науками, как биохимия, биоорганическая химия, фармакология и фармацевтическая химия. Основные понятия Основные понятия, используемые в медицинской химии — это мишень и лекарство. Мишень — это макромолекулярная биологическая структура, предположительно связанная с определенной функцией, нарушение которой приводит к заболеванию и на которую необходимо совершить определенное воздействие. Наиболее часто встречающиеся мишени — это рецепторы и ферменты. Лекарство — это химическое соединение (как правило, низкомолекулярное), специфически взаимодействующее с мишенью и тем или иным образом модифицирующее клеточный ответ, создаваемый мишенью. Если в качестве мишени выступает рецептор, то лекарство будет, скорее всего, его лигандом, то есть соединением, специфическим образом взаимодействующим с активным сайтом рецептора. В отсутствие лиганда рецептор характеризуется собственным уровнем клеточного ответа — так называемой базальной активностью. По типу модификации клеточного ответа лиганды делят на три группы (см. рис. 3):
Агонисты увеличивают клеточный ответ; Нейтральные агонисты связываются с рецептором, но не изменяют клеточный ответ по сравнению с базальным уровнем; Обратные агонисты, или антагонисты понижают клеточный ответ. Степень взаимодействия лиганда с мишенью измеряют аффинностью, или сродством. Аффинность равна концентрации лиганда, при которой половина мишеней связана с лигандом. Биологической же характеристикой лиганда является его активность, то есть та концентрация лиганда, при которой клеточный ответ равен половине максимального. Клинические исследования Медицина — это область, в которой ни в коем случае не следует спешить. В особенности, если речь идет о разработке новых лекарственных препаратов. Достаточно вспомнить историю с препаратом Талидамидом, разработанным в конце 50-х в Германии, применение которого беременными женщинами приводило к рождению детей с врожденными пороками конечностей, вплоть до их полного отсутствия. Этот побочный эффект не был вовремя выявлен во время клинических исследований в силу недостаточно тщательного и аккуратного тестирования. Поэтому в настоящее время процедура тестирования лекарств достаточно сложна, дорога и требует значительного времени (2-7 лет тестирования в клинике и от 100 миллионов долларов на одно соединение-кандидат, см. рис. 7).
Прежде всего, еще до поступления в клинику, препараты исследуются на токсичность и канцерогенность, причем исследования должны проводиться, кроме систем in vitro, как минимум на двух видах лабораторных животных. Токсичные препараты, само собой, в клинику не попадают, за исключением тех случаев, когда они предназначены для терапии особо тяжелых заболеваний и не имеют пока менее токсичных аналогов. Кроме того, препараты подвергаются фармакокинетическим исследованиям, то есть тестируются на такие физиологические и биохимические характеристики, как поглощение, распределение, метаболизм и выведение (по-английски обозначается аббревиатурой ADME — Absorption, Distribution, Metabolism and Extraction). Биодоступность, например, является подхарактеристикой введения препарата в организм, характеризующая степень потери им биологических свойств при введении в организм. Так, инсулин, принимаемый перорально (через рот), имеет низкую биодоступность, так как, будучи белком, расщепляется желудочными ферментами. Поэтому инсулин вводят либо подкожно, либо внутримышечно. По этой же причине часто разрабатывают препараты, действующие аналогично своим природным прототипам, но имеющие небелковую природу. Юридически процесс клинических исследований новых препаратов имеет очень много нюансов, так как они требуют огромного количества сопроводительной документации (в сумме несколько тысяч страниц), разрешений, сертификаций и т.д. Кроме того, многие формальные процедуры сильно разнятся в разных странах в силу различного законодательства. Поэтому, для решения этих многочисленных вопросов, существуют специальные компании, принимающие от крупных фармацевтических компаний заказ на проведение клинических испытаний и перенаправляющие их в конкретные клиники, сопровождая весь процесс полной документацией и следя, чтобы никакие формальности не были нарушены. Перспектива драг-дизайна Очевидно, что драг-дизайн — это будущее фармакологической промышленности. Направленное конструирование новых лекарственных препаратов уже сейчас стало важнейшей частью современного общества, позволяя победить многие болезни, излечение которых ранее не представлялось возможным. В перспективе, новые наукоемкие приложения смогут поднять драг-дизайн на еще более высокий уровень, когда будут, наконец, побеждены такие тяжелые заболевания, как рак, СПИД, болезнь Альцгеймера и другие недуги человечества. Где расположены мишени?
Прежде всего, следует указать на мембранно-связанные рецепторы. Например, холинорецептор — это белковый комплекс, функции которого связаны с регуляцией клеточной мембраны для изменения ее проницаемости для катионов натрия, калия, кальция. Он находится на синаптической мембране, фрагменты которой обеспечивают возрастание проницаемости для Na+, K+, Са++ под действием агонистов типа карбахолина — соединения структурно близкого медиатору — ацетилхолину.
Антагонистом данных рецепторов является атропин, который может быть вытеснен с рецепторной поверхности ацетилхолином.
Активные центры различных ферментов часто выступают в роли рецепторов. Так, известный препарат, применяющийся в глазной практике для сужения зрачка и понижения внутреннего давления при глаукоме — физостигмин обратимо взаимодействует с рецептором на ацетилхолинэстеразе.
Ацетилхолинэстераза — это фермент, гидролизующий эфирные связи и специфичный по отношению к ацетилхолину. С тем же ферментом взаимодействует ингибитор холинэстеразы прозерин. Он вытесняет ацетилхолин из активного центра фермента (структурное сходство определяется наличием в обоих соединениях четвертичной триметиламмониевой группировки и эфирной группы на близком расстоянии) и проявляет прямой эффект на мышечный холинорецептор.
Другим важным примером рецептора на ферменте является место специфического связывания пара-аминобензойной кислоты (ПАБ) на дигидрофолатсинтетазе. ПАБ является природным субстратом этого фермента, из которой в организме синтезируется дигидрофолиевая кислота. Нарушение этого синтеза структурно близкими сульфамидами является основой их антибактериальной активности.
Ингибирование фермента РНК-полимеразы — основа действия антибиотика рифампицина. Здесь так же действие обусловлено связыванием с препарата с рецепторным участком фермента.
Блокада рецепторов, находящихся на ферменте дигидрофолатредук-тазе обусловливает эффекты противомалярийного препарата хлоридина и антибактериального средства триметоприма
Наконец, еще один пример взаимодействия лекарственного средства с рецепторным активным участком фермента — бензилпеницил-лин проявляет свою антибактериальную активность за счет блокады по-лимеразы, участвующей в построении клеточной стенки бактерий.
Теперь остановимся на рецепторах, являющихся частью нуклеиновых кислот. К лекарствам, имеющим места рецепторного связывания на нуклеиновых кислотах, относятся аналоги пуриновых и пиримидиновых оснований, которые способны встраиваться между цепями ДНК (например, идоксуридин) или РНК (например, 8-азагуанин) оказывая антибактериальное и противовирусное действие.
Плоская молекула биологически активного вещества способна вклиниться между парами оснований дезоксирибонуклеиновой кислоты (ДНК), образуя комплекс, который стабилизируется силами Ван-дер-Ваальса и ионными связями. При этом затрудняется возможность расплетания двойной спирали, нарушается синтез бактериальных ДНК и РНК с участием полимераз. Примечательно, что различные биологически активные вещества могут связываться с различными рецепторными системами, и в то же время обеспечивать сходные физиологические ответы. Так, например, гипотензивный эффект (снижение артериального давления) может быть вызван апрессином, воздействующим на периферические сосуды, р-адреноб-локаторами (например, анаприлином — блокада симпатических нервных окончаний), гексонием, блокирующим симпатические ганглии, ингибитором ангиотензин-конвертирующего фермента — капотеном, донором оксида азота — молсидомином и катапрессаном (клофелином), действующим на адренорецепторы.
Медиаторы Рецепторные системы организма изначально приспособлены для взаимодействия с эндогенными соединениями, такими как нейромедиаторы, оказывающими принципиальное влияние на протекание большинства жизненно важных процессов. Связывание именно с этими соединениями является наиболее эффективным, и поэтому значительная часть поисковых исследований направлена на синтез веществ, имеющих ту или иную степень структурного сходства с нейромедиаторами. К числу нейромедиаторов относится упоминавшийся ранее ацетилхолин, принимающий участие в передаче нервного возбуждения в центральной нервной системе, вегетативных узлах, окончаниях парасимпатических и двигательных нервов Ацетилхолин является медиатором (передатчиком) нервного возбуждения. Рецепторные системы, взаимодействующие с ацетилхолином, называются холинорецепторами.
Главным медиатором передачи нервного возбуждения с симпатических нервных окончаний на эффекторные клетки (адренергическая система, адренорецепторы) является норадреналин.
Упомянем такие нейромедиаторы, как дофамин (4), являющийся к тому же предшественником норадреналина и адреналина в биосинтезе, серотонин (5), взаимодействующий со специфическими серотониновыми рецепторами в центре и на периферии и гистамин (6), рассматривающийся сейчас как «медиатор» воспаления и аллергии и играющий роль нейромедиатора в ЦНС.
К числу нейромедиаторов относятся аминокислоты: гамма-амино-масляная кислота (ГАМК) (7) и глицин (8) (тормозные медиаторы) и глутаминовая (9) и аспарагиновая (10) кислоты (возбуждающие медиаторы, стимулирующие передачу возбуждения в синапсах ЦНС)
Список медиаторов можно долго продолжать. Важно то, что та или иная степень структурного сходства с медиаторами — один из важных путей изыскания веществ, тропных к рецепторным системам и являющихся либо блокаторами активности медиаторных соединений - антагонистами, либо агонистами — веществами, связывающимися с рецепторами и оказывающими физиологические эффекты, сходные с эффектами медиаторов. Антиметаболиты
Как обсуждалось выше – препараты в организме разлагаются под действием ферментов. Вещества блокирующие действия ферментов называются антиметаболитами. Такие вещества должны быть, также, как истинные субстраты, комплементарны тем активным участкам ферментов, на которых происходят процессы метаболизма. Ферменты состоят из коферментов и апоферментов. Коферменты — низкомолекулярные органические соединения биологического происхождения, необходимые для действия ферментов. Это переносчики электронов или различных группировок, таких как фосфатные, ацильные и другие группы, активаторы молекул субстратов. Апоферменты – это специфические белки, которые образуют с коферментами комплексы. Основной функцией ферментов является ускорение химических реакций (например: энергия активации разложения перекиси водорода каталазой снижается с 18 до 2 ккал/моль, т. е. скорость процесса Н2О2 —>Н2О + О увеличивается под действием фермента в 1,6 * 1011 раза). Причины такой каталитической активности — это образование максимально энергетически выгодного переходного состояния за счет изменения конформаций субстрата и фермента, а также близости реагирующих группировок, концентрирующихся на активном центре фермента, где к тому же организуется практически безводная среда с низкой диэлектрической постоянной, способствующая взаимодействию ионных или дипольных группировок. Соединения, подходящие с точки зрения подавления ферментативной активности, как уже указано выше, должны быть структурно сходными с субстратами или коферментами и, таким образом, действовать как антиметаболиты. Существуют определенные эмпирически найденные (и в ряде случаев обоснованные теоретически) правила поиска антиметаболитов — поиска, весьма перспективного с точки зрения создания новых эффективных лекарственных препаратов. Некоторые из этих подходов могут быть кратко изложены таким образом: превращение метаболита — например, карбоновой кислоты в антиметаболит возможно при замене карбоксильной группы на сульфамидную, бензольное кольцо может быть заменено на пиридиновое или тиофеновое, вместо ароматических или гетероциклических аминопроизводных рекомендуется использовать соответствующие оксипроизводные.
Замена атомов водорода и метальных групп соответственно на атомы фтора и хлора также зачастую оказывается весьма эффективной.
Между метаболитами и антиметаболитами имеются конкурентные отношения. Для каждой пары может быть выведен индекс ингибирования — отношение концентрации антиметаболита к концентрации метаболита, при котором достигается 50 %-е ингибирование. Вообще говоря, к взаимоотношениям антиметаболит-метаболит вполне применимы обычные соотношения для ингибирования ферментов.
Где Ki – константа ингибирования [E] – концентрация фермента [I] – концентрация ингибитора [EI] – концентрация комплекса Наиболее впечатляющий пример получения антиметаболитов относится к ингибированию синтеза дигидрофолиевой кислоты сульфамидными препаратами. Как видно из структуры дигидрофолиевой кислоты, центральным компонентом ее формулы является остаток пара-аминобензойной кислоты (ПАБ)
Дигидрофолиевая кислота играет жизненно важную роль в живых организмах как у млекопитающих, так и у бактерий. Млекопитающие получают ее вместе с пищей, в то время как бактерии синтезируют ее на основа ПАБ на ферменте дигидрофоласинтетазе. Сульфаниламиды близки по структуре, электронным свойствам и размерам молекулы к ПАБ.
Они ингибируют синтез дигидрофолиевой кислоты, что приводит к гибели патогенных бактерий, не затрагивая жизненно важных функций организма млекопитающих. Дигидрофолиевая кислота далее на дигидрофолатредуктазе восстанавливается до тетрагидрофолиевой кислоты
Из антиметаболитов дигидрофолиевой кислоты наиболее известен противоопухолевый препарат метотрексат
Большинство простейших организмов не способно синтезировать пурины и должны получать их от организма хозяина и иметь ферментные системы, пригодные для их переработки. Отсюда — поиск соответствующих антиметаболитов. В качестве примера можно привести структурный аналог гипоксантина — аллопуринол.
И, наконец, укажем на антиметаболит гамма-аминомасляной кислоты (ГАМК) — 3-винил-гамма-аминомасляную кислоту, которая повышает содержание ГАМК в мозге за счет ингибирования фермента ГАМК-трансаминазы, субстратом которого является сама гамма-амино-масляная кислота.
Способы синтеза виагры В настоящее время известны три коммерческих ингибитора фосфодиэстеразы (V) – силденафил, варденафил и тадалафил.
Первый коммерческий PDE (V) – Силденафил цитрат, являющийся активным ингредиентом «Виагры» был зарегистрирован в сентябре 1997 года. Поскольку это был первый орально вводимый препарат для лечения MED, то уже через 6 месяцев после регистрации в FDA он получил патент и был выпущен на рынок. Два других коммерческих PDE (V) – ингибитора, были на утверждении в FDA более длительное время. Эти два препарата (варденафил и тадалафил) были выпущены на рынок в США во второй половине 2003 года. Как видно из рис. 1, Варденафил является структурным аналогом Силденафила, в то время как Тадалафил является принципиально иной молекулой, базирующейся на тетрагидро-β-карбомине. Со времени выхода «Виагры» на рынок в 1998 году спрос на коммерческие ингибиторы PDE (V) постоянно растет (рис. 2) [49] и вероятнее всего, что эта тенденция будет продолжаться и в дальнейшем.
Рис.2 Данные по мировой продаже препаратов-ингибиторов фосфодиэстеразы (v) Впервые сиденафил был синтезирован по схеме:
Первоначальные условия (1990): (i) Step 1, T, SOCI2 ацетон, затем NH3 (водный) (78 %), (ii) Step 2a SnCI2, T, этанол (94 %) (iii) Step 2b (COCI)2, CH2CI2 перегонка (89 %), (iv) Step 2с, ТЭА, CH2CI2, 25°C, хроматографическая очистка (40%) (v) Step 3, NaOH, этанол, H2O2; затем экстракция смесью CH2CI2: метанол, затем хроматографическая очистка (72 %) (vi) Step 4, CISO3H, затем выделение в воду, затем экстракция смесью CH2CI2: метанол. (vii) Step 4, N-метилпиперазин, этанол (viii) Step 5, образование соли лимонной кислоты. Выход продукта в расчете на 1 – 9,8 %
Альтернативные пути синтеза: Такой метод предложила индийская компания Oprug [62]. (Синтез через реакцию Фриделя – Крафтса). Сульфамоилхлорид 19 был получен взаимодействием N-метилпиперазина и SO2Cl2. Далее этот сульфохлорид использован для взаимодействия с пиразолпиримидиноном 14 по реакции Фриделя – Крафтса.
Схема 12. Реакция Фриделя – Крафтса Первым делом необходимо было синтезировать несколько килограммов вещества для предварительного исследования токсичности. Четыре изменения: 1. Использование стехиометрических количеств SOCI2 в растворителе, а не SOCI2 как растворитель на стадии (i). Это безопаснее и имеет меньшее экологическое воздействие. 2. Удаление восстановления SnCl2 на стадии (ii) Олово – тяж. Металл, 3. Использование SOCI2, а не оксалилхлорида на стадии (iii). Эта замена дала усовершенствование безопасности рабочего персонала, предотвращая эмиссию угарного газа. 4. Устранение перекиси водорода из синтеза. Водородный пероксид причиняет ожоги на контакте с кожей и – пожароопасен при транспортировке и особенно в контакте с органическим материами.
Химики используют восстановление хлоридом олова, в случае когда обычное восстановление водородом не было эффективно. Исследование показало, что неудача гидрирования была по причине следов соединений серы, от предыдущей стадии. Строгий контроль этих примесей, например, применение стехиометрического количества SOCI2 позволило избавиться от олова, и заменить эту стадию каталитическим восстановлением водородом. Каталитические гидрирования - среди самых чистых экологических химических стадий. В реакции преврашения nitro группы в амин, единственный побочный продукт - вода и есть возможности для регенерации растворителя и катализатора. Первоначально реакцию циклизации (4) в (5) проводили в водном растворе гидроокиси натрия и водородного пероксида с 72%-ом выходом. Исследования показали, что с использованием т-бутилата К в т-бутиловом спирте можно получить 5 с выходом 100% без заметного кол-ва примесей. Эта чистая реакция с высоким выходом стала первым шагом в разработке коммерческого маршрута.
Т.о.: Оптимизированные условия (1994) Optimised Med Chem Process (1994) (i) Step 1, SOCI2, толуол 50-60°C (92%), (ii) Step 2a H2, Pd / C, этилацетат (100%), (iii) Step 2b, SOCI2 ДМФА (cat), этилацетат (100 %) (iv) Step 2с, пиридин, этилацетат (84 %) (v) Step 3, третбутилат калия. Третбутиловый спирт (100%) (vi) Step 4, CISO3H, затем выделение в воду, затем экстракция CH2CI2 (vii) N-метилпиперазин, толуол, (71 %), перекристаллизация из 2-бутанона (80%) (viii) Step 5, лимонная кислота, ацетон (91%), перекристаллизация из водного ацетона (90 %).
Суммарный выход 35,9%
Главные идеи коммерческого пути синтеза представлены на схеме:
стратегические преимущества коммерческого маршрута были следующие:
· Большая выход · Чистая реакция cyclisation - теперь в конце пути. Потенциально опасные материалы теперь используются в начале синтеза. · Экологические проблемы с использованием HOSO2Cl (то есть большие объемы водных кислых сливов, требующих neutralisation и повышенный уровень гидролиза продукта на стадии выделения сульфохлорида в воду, уменьшены, за счет перевода реакции
Дальнейшие улучшения были предложены в рамках этой схемы
1. Использование 1 моль SOCI2 вместе с HOSO2Cl на стадии СХ ЭБК для полной конверсии СК в СХ 2. Низкая Тпл. ЭБК (19-20) позволяет использовать ее расплав и использовать сравнительно меньшие кол-ва ХСК. 3. Для упрощения полученный влажный СХ суспензируется в воде и так реагирует с с N-methylpiperazine. После реакции рН доводится до 7 и сульфамид отфильтровывается. Т.О. на этой стадии не используются органический р-ли. 4. Использование толуола в кач-ве р-ля на 1-й стадии позволило снизить к-во SOCI2 до 1,2-1,6 моль/моль. Толуол рециклизуется. 5. Восстановление в ЭА водородом. 6. Активация к-ты 6 с пом. КДИ. Преимущества КДИ · чистая химия и высокое качество продукта. · Позволяет объеденить все три реакции гидрирование, активация и · Единый растворитель, который используется (ацетат этила), · Не используются хлорсодержащие реагенты. · Высокий выход, (90 %) на три стадии
Полученый продукт циклизуют тБК в тБС. Процесс ведут при высоких конц. Для уменьшения к-ва отходов. По оконч. Р-ии добавляют воду, рН доводят до изоэлектрической точки и осадок сиденафила отфильтровывают. Продуктом явл. Лимоннокислая соль. Был разработан метод получения СЦ сочетающий высокий выход (98-100%) и высокое качество. Т.О. Условия коммерческого метода синтеза (1997): (i) Step 1, SOCI2, DMF (cat.), Т толуол; NH3 (водный) (92%) (ii) Step 3a, H2 Pd/C этилацетат (100%) (iii) Step 2, CISO3H, SOCI2 25°C; (iv) Step 2, N-метилпиперазин, вода, 25°C затем нейтрализация (v) Step 3b, 6 + CDI, этилацетат, затем + 3 (90 %) (vi) Step 4, третбутилат калия. Третбутиловый спирт (92 %) (vii) Step 5, лимонная кислота, 2-бутанон (99 %).
1997 г - Суммарный выход 75%
Как только стадии были разработаны, начались исследования по рециклизации растворителей, используемых в синтезе. Рециклизация толуола, этилацетата и 2-бутанона была введена в 1998, году, когда Viagra ™ был запущен на рынок. Как только стадии были разработаны, начались исследования по рециклизации растворителей, используемых в синтезе. Рециклизация толуола, этилацетата и 2-бутанона была введена в 1998, году, когда Viagra ™ был запущен на рынок. Использование растворителей на разных стадиях разработки процесса дается в рис. 1.
Рис. 1 количество органической траты(отходов), произведенной sildenafil солью лимонной кислоты обрабатывает в различных пунктах(точках) времени.
Из специфического примечания - полное устранение хлорированных растворителей и устранения очень летучих растворителей типа диэтилового эфира, хлорида метилена, метанола и ацетона. Устранение этих очень летучих растворителей уменьшает атмосферное загрязнение, как будет отмечено позже. Дальнейшее усовершенствование синтеза (описанного в Схеме 3) было заменой т-бутанола. Этот растворитель полностью смешивается с водой в любых соотношениях и трудно восстанавливается для повторного использования. Есть перспективы для улучшенной экологических характеристик процесса за счет перехода от т-бутанола к другому растворителю, который может быть восстановлен. Этот новый процесс был исследуется и оптимизируется. (данные на 2003 г.) и после введения его в эксплуатацию расход растворителей составит 4л/кг.
Коммерческий синтез Силденафила представлен на схеме 1 [50]. Ключевым соединением для этой схемы является нитропиразол 4, синтез которого описан в разделе 2.4. Другое ключевое соединение – сульфамид 5. Его синтез осуществляется путем последовательного сульфохлорирования исходной кислоты и амидирования сульфохлорида. Взаимодействие, приводящее к соединению 6 осуществляется когда вещества находятся в виде раствора в этилацетате в одной реакционной массе, и при этом нитропиразол подвергается каталитическому восстановлению в присутствии КДИ.
Схема 1. Коммерческий синтез Силденафил цитрата
Альтернативные пути синтеза: Во-первых конденсация альдегида 7 с аминопиразолом 8 в кипящем толуоле приводит к получению дигидросилденафила 9. Выход по данной реакции составляет 52 – 95 %. Выделение целевого продукта осуществляется путем отгонки толуола азеотропной дистилляцией и в зависимости глубины дистилляции изменяется выход. Дигидросилденафил 9 далее окисляют до Силденафила: а) с использованием катализатора Pd/С в присутствии малых количеств трифторуксусной кислоты при высокой температуре; б) либо гидросульфата натрия. Химия этой схемы описана в работах [52, 53] в случае, когда, окисляющим агентом является гидросульфат натрия, общий выход 1 в расчете на аминопиразол составляет 85 %. Условия получения 9: кипящий толуол (52 %) или кипящий толуол с последующей азеотропной отгонкой (92 %).
Схема 2. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 1)
Условие получения 1 из 9: Pd/С, трифторуксусная кислота, толуол (200 °С, 84 %) или NaHSO3, кипящий диметилацетальдегид – 90 %.
Построение пиримидинового фрагмента может осуществляться на основе нитрила 12, либо через амидин 10, либо через иминоэфир 11. Эти подходы и, соответственно, экспериментальные процедуры, были описаны в работе сотрудников фирмы Pfizer [54, 55]. Амидин 10 был получен из нитрила 12 взаимодействием его с хлорметилалюминием (полученным на основе хлорида аммония и триметилалюминия).
Схема 3. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 2)
Условия получения амидина 10: гексан, Me3Al, NH4Cl, толуол, исходное – 12, сначала 12,5 °С, затем 80 °С (58 % выход). Иминоэфир 11 получен из нитрила 12 по реакции Пиннера.
Схема 4. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 3)
Условие этой реакции: этанол, пропускание газообразного HCl, 0 °С (выход 48 %).
Третий синтез – это синтез через интермедиат 13, содержащий кислотную функциональную группу в пиридиноновом цикле
Схема 5. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 4)
Циклизация интермидиата-кислоты протекает с использованием тионилхлорида и приводит к лактону 13а, который далее взаимодействует с аммиаком, приводит к пиразолпиримидинону 1 (Силденафилу). Данные процедуры описаны в работе [56]. Условия получения 1 из 13. Синтез 13а из 13: кипячение в SO2Cl2 с последующей отгонкой SOCl2. Синтез 1 из 13а в этаноле обработка аммиаком. Следующий синтез на основе нитропиразола 4.Этот подход описан в работе [57]. Соединение 15, содержащее атом F в качестве радикала R было получено при взаимодействии 2 – фторбензоилхлорида с пиразолом 4 (выход 65 %) с последующим сульфохлорированием и сульфамидированием (75 %). Далее нитрогруппа была восстановлена с помощью SnCl2 и полученный амин принял участие в реакции циклизации. На заключительной стадии (получение 1 из 15) атом F был замещен на группу OEt.
Схема 6. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 5)
Условия получения 1 из 15: восстановитель - SnCl2, EtOH, 65 °С.
Другой вариант, также как и основная, коммерчески реализуемая схема, приводит к интермедиату 6, циклизация которого дает целевой Силденафил. Однако получение интермедиата 6 основано не на 2-этоксибензойной кислоте (в качестве исходного), а на его амидном производном 16, которое сначала подвергается сульфохлорированию и амидизации сульфохлорида, а затем уже и циклизации до Силденафила.
Схема 7. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 6)
Схема 7 описана в работе [58]. Условия циклизации 6 в 1 описаны выше – KOBu, кипячение в бутиловом спирте в течение 6 – 8 часов с выходом 95 %.
Последний вариант синтеза Силденафила, заключительной стадией которого, является реакция циклизации представлен на схеме 8.
Схема 8. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 7)
Этот вариант также является коммерчески оправданным, так как в качестве исходного используется 2-хлор(фтор)бензойная кислота, которая является более дешевой, чем этоксибензойная кислота. На последней стадии данного процесса, при использовании этанола в качестве растворителя и сильного основания для активирования циклизации, происходит одновременное протекание циклизации и замещение уходящей группы. В случае, когда Х=F, выход 1 в расчете на исходный реагент составляет 100 % [59].
Антибиотики Антибиотики—вещества природного происхождения, обладающие выраженной биологической активностью. Они получаются из растений, микробов, животных тканей, а также синтетическим путем.
Среди антибиотиков значительное место занимают пенициллины. Пенициллины оказывают выраженный эффект в отношении растущих микроорганизмов (бактерицидное действие), они поражают бактерии в фазе роста, ослабляя их клеточные стенки, а, учитывая, что для бактерий характерно необычайно высокое внутреннее давление, это приводит к разрыву клеточной стенки и уничтожению бактерии. Следует отметить, что бактерии резко отличаются от всех живых организмов, в частности, необычным составом клеточных стенок, основным компонентом которых является ацетилмураминовая кислота
Создание полимера протекает ферментативно. пенициллины выступают в качестве ацилирующих агентов по отношению к транспептидазной части фермента, причем ацилирование происходит за счет легко раскрывающегося Р-лактамного цикла. Открытие Флемингом плесени Penicillinum позволило с 1941 г. наладить ее получение в количествах, достаточных для испытания на людях. Дальнейшие усилия исследователей разных стран привели к созданию высокоэффективных штаммов пенициллинов, разработке рациональных условий глубинной ферментации, выделения и очистки пенициллинов, способов и условий их применения
Было установлено, что ядро пенициллина состоит из р-лактамтиазолидиновой бициклической системы, образовавшейся конденсацией аминокислот dl-валина и 1-цистеина.
Отдельные пенициллины отличаются друг от друга различными остатками R, от строения которых в основном зависит их различное биологическое действие. С расшифровкой строения природных пенициллинов начались работы по синтезу и модификации их структур Рядом исследователей было показано, что добавление фенилуксусной кислоты в ферментационную среду приводит к повышению выхода бензилпенициллина. Это открытие привело к разработке метода биосинтеза, заключающегося в добавлении в питательную среду различных органических кислот—предшественников, используемых плесневыми грибками для построения бокового радикала. | ||||
|
| Поделиться: |



 Для разработки лекарства важна проблема локализации мишеней, то есть, вопрос, где расположены биологические структуры, подверженные действию лекарств.
Для разработки лекарства важна проблема локализации мишеней, то есть, вопрос, где расположены биологические структуры, подверженные действию лекарств.

 Атропин
Атропин физиостигмин
физиостигмин Прозерин
Прозерин