Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Элементы химической термодинамики, термодинамики растворовСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Лекция №1 Элементы химической термодинамики, термодинамики растворов И химической кинетики Значение химии в медицине · 78 элементов входят в состав живых организмов. · 44 элемента составляют лекарственные препараты. · Изотопы 38 элементов используются в диагностике и радиотерапии различных заболеваний. · Более 70 элементов входят в состав материалов, применяемых для изготовления медицинской аппаратуры, приборов, инструментов, перевязочных средств, искусственной крови, различных протезов, зуботехнических материалов и др. · В организме человека реализуется около 100 тысяч химических превращений. · Живая клетка функционирует по строгим законам химии. · Более 75 % лекарственных средств производит химико-фармацевтическая промышленность. Задача, состоящая перед медиками в ближайшее время, предупреждать, а не лечить болезни. Чтобы стать высококвалифицированным специалистом нужно помнить высказывание М.В. Ломоносова: «… Медик без довольного познания химии совершенен быть не может… От одной химии уповать можно на исправление недостатков лечебной науки» Термодинамика – наука, изучающая общие законы взаимного превращения одной формы энергии в другую. К настоящему времени термодинамика содержит два основных раздела: 1. Равновесная термодинамика (термодинамика изолированных систем) В основном разработана в середине 19-го – начале 20-го века и содержит три закона – три «Начала»: · -в середине 19-го века Ю. Р. Майером, Дж. Джоулем и Г. Гельмгольцем был сформулирован первые закон термодинамики - «Первое начало термодинамики». · - в 1850 году Р. Клаузиусом, и независимо от него в 1851 году У. Томсоном было сформулировано «Второе начало термодинамики». · -в 1906 году В. Нернст сформулировал «Третье начало термодинамики». 2. Неравновесная термодинамика(термодинамика открытых систем) Разработана в 20-м веке. Содержит два основных подраздела: · -слабо неравновесную термодинамику, основы которой разработаны в 1931 Л. Онсагером; · -сильно неравновесную термодинамику, в основном разработанную Г. Хакеном, И. Пригожиным и Р. Томом в середине 20-го века. Первой работой в области неравновесной термодинамики в биологии является опубликованная в 1935 году книга Э.Бауэра «Теоретическая биология», в которой был сформулирован «Всеобщий закон биологии». 1.2. Основные термины и положения термодинамики При изучении термодинамики пользуются определенными понятиями. Система – это совокупность материальных объектов (тел), ограниченных каким-либо образом от окружающей среды. Это может быть раствор любого вещества, организм животного, состоящий из взаимодействующих элементов- органов. Элементами называются части, обладающие определенными свойствами. В случае биологических объектов имеем ряд: Биосфера – биоценоз – популяция – организм – орган – ткань – клетка – органелла - молекула. При взаимодействии термодинамической системы с окружающей средой происходит обмен энергией. Возможны два способа передачи энергии. Упорядоченная форма передачи энергии, которая связана с изменением внешних (объема и давления) параметров состояния системы, называют работой. Неупорядоченную форму передачи энергии называют теплотой. В термодинамике под термином «работа» чаще понимают не сам процесс, а количество передаваемой при этом энергии. Работу, производимую системой над окружающей средой, принято считать положительной, работу, производимую над системой принято считать отрицательной. Под термином «теплота» понимают не сам процесс, а количество передаваемой энергии Q. Q положительна, если система получает некоторое количество энергии в форме теплоты, при передаче энергии в противоположном направлении величину Q считают отрицательной. Единица измерения в СИ – джоуль (Дж). Термодинамические системы могут быть следующими: • Гомогенная – система, в которой каждое свойство ее (параметр) имеет одно и то же значение во всех точках объема или меняется плавно от точки к точке. • Гетерогенная – такая система, которая состоит из нескольких гомогенных систем, отделенных друг от друга поверхностью разделяя фаз, на которой свойства меняются скачком. Организм человека состоит из многофазных систем – ткани, кровь, лимфа, слюна и др. •Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией в форме работы или теплоты. • Закрытая (замкнутая) система – система, которая может обмениваться с окружающей средой лишь энергией и не может обмениваться веществом; • Открытая система – система, которая обменивается с окружающей средой и энергией, и веществом. • Адиабатная система – нет теплообмена с окружающей средой. Живые организмы являются открытыми системами: организм человека за 40 лет жизни потребляет в среднем 40 т воды, 6 т пищи и около 12 млн л кислорода. Постоянный обмен между системой окружающей средой составляет материальную сущность жизнедеятельности - метаболизм. Состояние любой термодинамической системы характеризуется двумя группами параметров: Интенсивными термодинамическими параметрами (давление, температура и др.), не зависящими от массы или числа частиц в системе; Экстенсивными термодинамическими параметрами (общая энергия, энтропия, внутренняя энергия), зависящими от массы или числа частиц в системе. Изменение параметров термодинамической системы называется термодинамическим процессом. Различают - изотермические (при постоянной температуре, Т = соnst), - изобарные (при постоянном давлении, Р = соnst), - изохорные (при постоянном объеме, V = соnst), - адиабатические процессы (без теплообмена с окружающей средой). Энергию системы (W) можно представить как совокупность двух частей: зависящую от движения и положения системы как целого (W ц) и не зависящую от этих факторов (U).
Вторую составляющую этой совокупности U называют внутренней энергией системы. Она включает энергию теплового движения частиц, а также химическую и ядерную энергию, определяющую поступательное, колебательное и вращательное движение молекул, внутримолекулярное взаимодействие и колебание атомов, энергию вращения электронов. Внутренняя энергия в свою очередь разделяется на свободную энергию и связанную энергию. Свободная энергия (G) – та часть внутренней энергии, которая может быть использована для совершения работы. Связанная энергия (W св) – та часть энергии, которую нельзя превратить в работу.
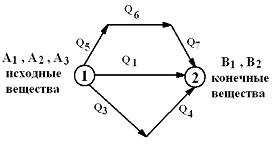
Общие сведения о равновесной термодинамике Тепловой эффект химических реакций, протекающих при постоянном давлении или при постоянном объёме, не зависит от числа промежуточных стадий, а определяется лишь начальным и конечным состоянием системы. Например, вещество АВ можно получить из А разными способами: 1) А + В = АВ (ΔН) 2) А + С = АС (ΔН1) АС + В = АВ + С (ΔН2) ΔН1 + ΔН2 = ΔН
В термодинамике принята следующая запись теплот химических реакций: С(тв) + О2(г) = СО2(г); ∆НР = -405,8 кДж; в термохимии: С(тв) + О2(г) = СО2(г) + 405,8 кДж. ∆Н0 указывает на стандартное состояние (Р = 1 атм, Т = 298 К). Реагирующие вещества берутся в том агрегатном состоянии и в виде той кристаллической модификации, которые наиболее устойчивы при данных условиях. В термодинамике считается положительной (+) теплота, поглощенная системой (эндотермический процесс), а в термохимии – выделяемая в результате реакции (экзотермический процесс). В термохимии пользуются понятием «теплота (энтальпия) образования вещества». Под теплотой образования понимают тепловой эффект реакции образования одного моль вещества из простых веществ. Существует также понятие «стандартная теплота образования вещества» - тепловой эффект реакции образования одного моль вещества из простых веществ в стандартных условиях ( ΔН0298) (при 298 К и 1 атм) Обычно теплоты образования простых веществ в стандартных условиях принимают равными нулю. Теплоты образования приводятся в справочниках. Большое значение закона Гесса заключается в том, что, пользуясь им можно вычислить неизвестную теплоту реакции путем комбинирования стехиометрических уравнений и теплот других реакций, изученных экспериментально. При этом необходимо сравнивать теплоты различных реакций в одних и тех же условиях. Пример: Определить тепловой эффект реакции на основе экспериментальных данных при 0°С и давлении 1 атм. · С + О2 = СО2; ∆НР,1 = -405,8 кДж · СО + ½ О2 = СО2; ∆НР,2 = -284,5 кДж ______________________________________ 3) С + ½ О2 = СО; ∆НР,3 -? (3) = (1) – (2) Проверка: С + О2 - ½ О2 – СО = СО2 - СО2; С + ½ О2 = СО. ∆НР,3 = ∆НР,1 - ∆НР,2 = -405,8 кДж – (-284,5 кДж) = 121,3 кДж. Для расчетов по закону Гесса часто пользуются теплотами сгорания органических соединений, которые можно довольно легко определить экспериментально. Теплота реакции рассчитывается по I следствию закона Гесса: Теплота реакции равна сумме теплот сгорания начальных участников реакции за вычетом суммы теплот сгорания конечных участников реакции с учетом стехиометрических коэффициентов.
где νн, νк - стехиометрические коэффициенты. Пример для реакции а А + b B→ d D * ∆Hреакц. = а ∆Hсгор(А) + b ∆Hсгор(B) - d ∆Hсгор(D). • Теплотой сгорания вещества называется тепловой эффект реакции сгорания его (1 моль) с образованием устойчивых продуктов (для органических веществ это СО2 и Н2О). Кроме теплот сгорания часто используют теплоты образования химических соединений. • Теплотой образования вещества называется теплота образования Простые вещества реагируют в виде той модификации и в том агрегатном состоянии, которые наиболее устойчивы при данных условиях. Теплоты образования лишь в редких случаях могут быть определены экспериментально, чаще теплоты образования рассчитывают на основе закона Гесса. Пример: а А + b B→ d D. ∆HР = ∆Hобр(D)· d = а ∆Hсгор(А) + b ∆Hсгор(B) - d ∆Hсгор(D);
Соединения, теплоты образования которых положительны, называются эндотермическими, а теплоты образования, которых отрицательны - экзотермическими. В целях сопоставления и использования для расчетов по закону Гесса теплоты огромного количества реакций (химических соединений) рассчитаны для стандартных условий:Р = 1 атм (101300 Па); Т = 298 К (25 °С) и сведены в таблицы стандартных величин ( Пользуясь стандартными значениями можно рассчитать тепловой эффект любой реакции (при стандартных условиях) по II следствию закона Гесса: Теплота реакции равна сумме теплот образования конечных веществ за вычетом суммы теплот образования начальных веществ с учетом стехиометрических коэффициентов.
Или для стандартных условий:
Пример: а А + b B→ d D ∆HР = d ∆Hобр(D) – (а ∆Hобр(А) + b ∆Hобр(B)) = d ∆Hобр(D) - а ∆Hобр(А) - b ∆Hобр(B). При этом величина и знак теплоты образования характеризуют устойчивость соединения в данных условиях. Например:
Чем меньше ΔН, тем более устойчиво соединение. При образовании NH3 выделяется тепло. Далее в приведённом ряду теплоты образования возрастают, и, следовательно, устойчивость соединений падает. А гидрид висмута разлагается при получении. NH4Cl = NH3 + HCl↑ Из справочника выпишем энтальпии веществ:
По этим данным можно рассчитать тепловой эффект хим реакции, пользуясь 2 следствием из закона Гесса. ΔН0298(реакции) = ΔН0298(HCl) + ΔН0298(NH3) - ΔН0298(NH4Cl) = 176,55 кДж/моль Тепловой эффект положителен, т.е. реакция эндотермическая, а, значит, чтобы разложить NH4Cl, его нужно нагреть. Если известен тепловой эффект реакции, то можно рассчитать и теплоту образования вещества, участвующего в реакции. Закон Гесса позволяет также рассчитывать теплоты образования неустойчивых соединений и тепловые эффекты реакций, которые нельзя осуществить экспериментально. На практике тепловой эффект реакций измеряют с помощью калориметра. Величина ΔН зависит от агрегатного состояния вещества, поэтому в термохимических уравнениях указывают агрегатное состояние веществ. Закон Гесса является следствием первого начала термодинамики и справедлив при постоянном объёме или постоянном давлении. Второе начало термодинамики Все процессы, протекающие в природе, самопроизвольно, то есть без затраты работы извне, имеют определенное направление. Так, самопроизвольно теплота переходит от нагретого тела к холодному, газ переходит из области большего давления в область низкого, в растворах самопроизвольно выравниваются концентрации и т.д. Из I-го закона т/д нельзя сделать вывод о возможности самопроизвольного протекания процесса. Поскольку все реальные процессы протекают в определенном направлении, то для изменения направления необходимо изменить условия протекания процессов. Все реальные процессы не являются равновесными и протекают с конечной скоростью, приближая систему к равновесию. В момент наступления равновесия процесс заканчивается. Неравновесный процесс, протекающий в направлении достижения равновесия без воздействия внешних условий, называется самопроизвольным (положительным). Обратный по направлению процесс, который не может протекать без внешних воздействий и удаляющий систему от равновесия называется несамопроизвольным (отрицательным). II закон термодинамики позволяет предсказать направление протекания процесса. Он имеет несколько формулировок: · Постулат Клаузиуса Единственным результатом любой совокупности процессов не может быть переход теплоты от менее нагретого тела к более нагретому. · Постулат Томсона Теплота наиболее холодного из участвующих в процессе тел не может служить источником работы. (Теплота не может полностью перейти в работу). · Вечный двигатель второго рода невозможен, т.е. невозможно построить такую машину, которая производила бы работу за счет тепла окружающей среды, не более нагретой, чем сама машина. Т.о., процесс превращения теплоты в работу является несамопроизвольным и для его проведения необходима специальная организация такого процесса (паровая машина). Пусть Т1– температура нагревателя, Т2 – температура холодильника. Пусть имеется цилиндр с поршнем и некоторым количеством вещества, которое называется рабочим телом (например, пар). Оно совершает циклическую последовательность процессов, периодически возвращаясь в исходное состояние. КПД такой машины - отношение совершенной работы к затраченной теплоте: η = Итальянский ученый Карно рассмотрел идеальную тепловую машину, в которой рабочим телом является 1 моль идеального газа, а все процессы совершаются идеально равновесно. ηидеальн = В идеальной машине Карно совершается максимальная работа, однако КПД составляет лишь около 40%, т.к. невозможно достичь температуры холодильника 0К. В реальной тепловой машине совершается меньшее количество работы и КПД существенно ниже. Энтропия Энтропия – функция состояния термодинамической системы, используемая во втором законе т/д для выражения через нее возможности или невозможности самопроизвольного протекания процесса (введена Клаузиусом). Изменение энтропии определяется отношением количества теплоты, сообщенного системе или отведенного от нее, к температуре системы:
где знак равенства относится к равновесному процессу, неравенства – к неравновесному. Т.о. в равновесном процессе: S = По изменению энтропии в изолированной системе можно предсказать т/д возможность протекания самопроизвольного неравновесного процесса. Если энтропия увеличивается (S > 0), то самопроизвольный неравновесный процесс возможен, если S < 0 – невозможен. Т.о. все самопроизвольные процессы в изолированных системах идут в сторону увеличения энтропии до достижения равновесия, где она будет иметь постоянное и максимальное значение. В современной термодинамике второе начало термодинамики изолированных систем формулируется единым и самым общим образом как закон возрастания особой функции состояния системы, которую Клаузиус назвал энтропией (S). Физический смысл энтропии состоит в том, что в случае, когда материальная система находится в полном термодинамическом равновесии, элементарные частицы, из которых состоит эта система, находятся в неуправляемом состоянии и совершают различные случайные хаотические движения. В принципе можно определить общее число этих всевозможных состояний. Параметр, который характеризует общее число этих состояний, и есть энтропия. Рассмотрим это на простом примере. Пусть изолированная система состоит из двух тел «1» и «2», обладающих неодинаковой температурой T 1 > T 2. Тело «1» отдает некоторое количество тепла Q, а тело «2» его получает. При этом идет тепловой поток от тела «1» к телу «2». По мере уравнивания температур увеличивается суммарное количество элементарных частиц тел «1» и «2», находящихся в тепловом равновесии. По мере увеличения этого количества частиц увеличивается и энтропия. И как только наступит полное тепловое равновесие тел «1» и «2», энтропия достигнет своего максимального значения. Таким образом, в замкнутой системе энтропия S при любом реальном процессе либо возрастает, либо остаётся неизменной, т. е. изменение энтропии Δ S =0. Знак равенства в этой формуле имеет место только для обратимых процессов. В состоянии равновесия, когда энтропия замкнутой системы достигает максимума, никакие макроскопические процессы в такой системе, согласно второму началу термодинамики, невозможны. Отсюда следует, что энтропия - физическая величина, количественно характеризующая особенности молекулярного строения системы, от которых зависят энергетические преобразования в ней. Связь энтропии с молекулярным строением системы первым объяснил Л. Больцман в 1887 году. Он установил статистический смысл энтропии (формула 1.6). Согласно Больцману (высокая упорядоченность имеет относительно низкую вероятность)
где k — постоянная Больцмана, P – статистический вес. k = 1.37·10-23Дж/К. Статистический вес Р пропорционален числу возможных микроскопических состояний элементов макроскопической системы (например, различных распределений значений координат и импульсов молекул газа, отвечающих определённому значению энергии, давления и других термодинамических параметров газа), т. е. характеризует возможное несоответствие микроскопического описания макросостояния. Для изолированной системы термодинамическая вероятность W данного макросостояния пропорциональна его статистическому весу и определяется энтропией системы:
Таким образом, закон возрастания энтропии имеет статистически-вероятностный характер и выражает постоянную тенденцию системы к переходу в более вероятное состояние. Отсюда следует, что наиболее вероятным состоянием, достижимым для системы, является такое, в котором события, происходящие в системе одновременно, статистически взаимно компенсируются. В связи с этим, введена ещё одна функция – ΔG – изобарно-изотермический потенциал (Энергия Гиббса) или свободная энергия Гиббса: ΔG = ΔH – TΔS где ΔH – изменение энтальпии, Т – абсолютная температура, ΔS – изменение энтропии. Если ΔG <0, процесс протекает самопроизвольно (экзэргонический процесс), если ΔG > 0, то процесс невозможен (эндэргонический процесс). ΔG <0, если ΔH<0, т.е. реакция экзотермическая, или ΔН > 0, но по абсолютной величине меньше T · ΔS (энтропийного фактора). Значение T · ΔS резко возрастает при высокой температуре и определяет направленность процесса. Этим объясняется изменение направленности некоторых реакций с повышением температуры. Третье начало термодинамики Третье начало термодинамики - закон термодинамики, сформулированный В. Нернстом в 1906 году (тепловой закон Нернста), согласно которому энтропия S любой системы стремится к конечному для неё пределу, не зависящему от давления, плотности или фазы, при стремлении температуры (Т) к абсолютному нулю. Третье начало термодинамики позволяет находить абсолютное значение энтропии, что нельзя сделать на основе первого и второго начал термодинамики. В классической термодинамике (первого и второго начал) энтропия может быть определена лишь с точностью до произвольной аддитивной постоянной S 0, что практически не мешает большинству термодинамических исследований, так как реально измеряется разность энтропий (S 0) в различных состояниях. Согласно третьему началу термодинамики при Т = 0 значение Δ S = 0. Макс Планк в 1911 году дал другую формулировку третьего начала термодинамики - как условие обращения в нуль энтропии всех тел при стремлении температуры к абсолютному нулю:
Отсюда S 0 = 0. Это даёт возможность определять абсолютное значения энтропии и других термодинамических потенциалов. Формулировка Планка соответствует определению энтропии в статистической физике через термодинамическую вероятность (W) состояния системы S = k ln W. При абсолютном нуле температуры система находится в основном квантово-механическом состоянии, для которого W = 1 (состояние реализуется единственным микрораспределением). Следовательно, энтропия S при Т = 0 равна нулю. В действительности при всех измерениях стремление энтропии к нулю начинает проявляться значительно раньше, чем может стать существенной при T → 0 дискретность квантовых уровней макроскопической системы, приводящая к явлениям квантового вырождения. Из третьего начала термодинамики следует, что абсолютного нуля температуры нельзя достичь ни в каком конечном процессе, связанном с изменением энтропии, к нему можно лишь асимптотически приближаться. Химическое равновесие Обратимые и необратимые реакции. Некоторые химические реакции прекращаются до того, как исходные вещества прореагируют полностью. Например, взаимодействие эквимолярных количеств водорода с йодом протекает при 350 °С до тех пор, пока не образуется 80 % HI от теоретически расчетного: H2+ I2↔ 2HI. Если HI нагревать при 350 °С, то происходит разложение на исходные Н2 и I2, однако процесс протекает таким образом, что образуется 10% Н2 и 10% I2, остальные же 80% HI не разлагаются: 2HI↔H2 + I2 Следовательно, при 350 °С осуществляются два процесса: прямая реакция, при которой из H2 и I2 образуется HI, и обратная реакция, в результате которой образовавшийся HI частично разлагается на исходные H2 и I2 Прямая и обратная реакции характеризуют состояние химического равновесия, т. е. системы, в которой не изменяется состав реагирующих веществ, если условия реакции остаются постоянными. Термодинамически химическое равновесие определяется как соотношение концентраций исходных веществ и продуктов реакции, при котором энтропия системы имеет максимальное, а изобарно-изотермический потенциал – минимальное значение. Реакции, протекающие одновременно в двух противоположных направлениях, называются обратимыми. В обратимой реакции при одинаковых условиях достигается состояние равновесия независимо от того, из каких веществ исходят. При записи подобных реакций вместо знака равенства пользуются противоположно направленными стрелками (↔). Рассмотрим взаимодействие магния с хлороводородом: Mg+2HCl=MgCl2 + H2↑. Эта реакция сопровождается образованием хлорида магния и водорода. Если попытаться осуществить обратную реакцию, т. е. пропускать водород через раствор MgCl2, то металлический магний и НС1 не получатся. Следовательно, данная реакция протекает только в одном направлении и поэтому называется необратимой. Изменение энергии Гиббса в химической реакции, протекающей в растворе, будет зависеть не только от температуры и давления, но и от количества каждого из веществ, входящих в состав раствора. Поэтому очень часто пользуются значением μi, представляющим собой частную производную энергии Гиббса по массе i-го вещества, при условии постоянства температуры и давления системы, а также масс остальных компонентов. Величина μi называется химическим потенциалом. Когда две системы с различными химическими потенциалами вступают во взаимодействие, то происходит выравнивание потенциалов за счет изменения массы (концентрации) веществ. Давление уравновешивается за счет изменения объема. Поэтому химический потенциал является движущей силой химических реакций. Если эти процессы происходят в гомогенной системе, то они приводят к установлению химического равновесия; в гетерогенной же среде имеет место фазовое равновесие. Константа химического равновесия. Самопроизвольное протекание обратимых химических реакций происходит до известного предела, т.е. до установления химического равновесия. Концентрации исходных веществ и продуктов реакции при этом остаются неизменными и называются равновесными. В условиях химического равновесия скорость прямой реакции равна скорости обратной. Например, для реакции mA + nB→pC + qD скорость прямой реакции v1= k1CАmCBn, скорость обратной реакции v2=k2CCpCDq. Как только обе скорости становятся одинаковыми, в системе устанавливается динамическое равновесие, и дальнейшее изменение концентраций всех участвующих в реакции веществ прекращается. Итак, в состоянии химического равновесия скорости прямой и обратной реакций равны: v1=v2, k1CАmCBn =k2CCpCDq. Kp= CCpCDq/ CАmCBn Это уравнение есть математическое выражение закона действующих масс, которому подчиняется система в состоянии равновесия: частное от деления произведения равновесных концентраций исходных веществ и продуктов реакции является величиной постоянной и называется константой равновесия Кр Константа равновесия — важнейшая характеристика химического взаимодействия, так как позволяет судить о полноте протекания реакции. Для необратимых процессов Кр→∞. Если же Кр=0, то это указывает на полное отсутствие химического процесса. Достижению химического равновесия иногда препятствуют некоторые факторы, причиной которых является специфика самого процесса. Проиллюстрируем это на примере реакции горения водорода: 2Н2 + О2 = 2Н2О При нормальных условиях газовая смесь водорода с кислородом может существовать бесконечно долго, так как в этих условиях они практически не реагируют между собой. Такое состояние не является равновесным, поскольку для протекания реакции между Н2 и О2 требуется внешнее воздействие. Если эту смесь поджечь, то произойдет мгновенное взаимодействие с образованием пара воды. Охлаждение этой системы до прежней температуры не возвращает ее в исходное состояние, так как новое состояние (образование воды) является термодинамически более устойчивым. Если состояние системы постоянно во времени, но при изменении внешних условий в системе происходит необратимый процесс, то такое состояние называется заторможенным (ложным) равновесием. Заторможенные равновесия характерны для твердофазных систем. Истинное химическое равновесие при отсутствии внешнего воздействия неизменно во времени, а после внешнего воздействия система может вернуться в прежнее состояние. При истинном химическом равновесии ΔG = 0, т. е. значение свободной энергии минимально. При заторможенном равновесии ΔG < 0. Константа равновесия и энергия Гиббса. Константа химического равновесия зависит от природы реагентов, от температуры и связана с изменением стандартной энергии Гиббса ΔG° химической реакции уравнением ΔG°= -RT lnK* Уравнение * позволяет по значению ΔG° определить К, а следовательно, и равновесные концентрации. Отсюда следует, что чем значительнее убыль энергии Гиббса, т. е. чем сильнее сдвинуто равновесие в сторону продуктов реакции, тем больше значение константы равновесия. При высоких отрицательных значениях ΔG° в равновесной смеси преобладают продукты взаимодействия. Если же ΔG° > 0, то в равновесной смеси преобладают исходные вещества. Объединяя уравнения ΔG = ΔH – T · ΔS и ΔG°= -RT lnK через величину ΔG, получим -RT lnK = ΔG° = ΔH0 – T · ΔS0 Это уравнение позволяет по значениям ΔH0 и ΔS0 вычислить константу равновесия и степень равновесного превращения. Очевидно, что константа равновесия в значительной мере зависит от температуры. Для эндотермических процессов повышение температуры соответствует увеличению константы равновесия, для экзотермических — ее уменьшению. От давления (если р не очень велико) константа равновесия не зависит. Смещение химического равновесия. Принцип Ле-Шателье. Состояние химического равновесия при изменении условий (температуры, давления или концентрации) может сместиться либо в сторону образования продуктов реакции, либо в сторону исходных веществ. Влияние, оказываемое на равновесную систему каким-либо внешним воздействием, можно предсказать, пользуясь принципом ЛеШателье (принципом подвижного равновесия): если на систему, находящуюся в равновесии, воздействовать извне, то в системе усилится то из направлений процесса, которое противодействует данному воздействию. Например, реакция окисления SO2 до SO3: 2SO2 (г)+О2 (r) = 2SO3 (г), ΔH = —396,1 кДж/моль Реакция образования SO3 сопровождается выделением теплоты, т. е. является экзотермическим процессом. Обратный процесс, т. е. разложение SO3 до исходных веществ, является эндотермическим. Если при установившемся равновесии повышать температуру, то это воздействие сместит равновесие в ту сторону, которая идет с поглощением теплоты. Таковым является разложение SO3. Так как при равновесии соблюдается условие ΔН = TΔS, то изменение температуры приводит к изменению и ΔН. При повышении температуры в системе усиливается действие энтропийного фактора (TΔS > 0), т. е. усиливается эндотермический процесс. При понижении температуры действие энтропийного фактора ослабевает и начинает преобладать экзотермический процесс. Влияние давления определяется изменением объема, которое происходит в ходе реакции. В данном примере по мере образования SO3 давление системы будет понижаться, поскольку из каждых двух молекул SO2 и одной молекулы О2, образуются только две молекулы SO3. Следовательно, при изменении давления равновесие будет смещаться в ту сторону, которая противодействует данному изменению; при увеличении давления равновесие смещается в сторону образования меньшего числа молекул газа, а уменьшение давления смещает равновесие в сторону образования большего числа молекул. Из этого следует, что в рассматриваемой равновесной системе увеличение давления сместит реакцию в сторону образования SO3. При изменении концентрации компонентов равновесной системы значение константы равновесия остается неизменным, однако само равновесие смещается, так как при этом создаются более благоприятные условия для протекания прямой пли обратной реакции. Увеличение концентрации SO2 или О2 (или одновременно и того и другого) сместит реакцию в сторону образования SO3, как процесса, приводящего к уменьшению концентрации SO2 и О2. Если по мере образования из реакционной среды удалять SO3, то равновесие тоже сдвинется вправо. Таким образом, если в реакционную смесь ввести избыток одного из исходных веществ, то равновесие смещается в сторону образования продуктов реакции. Аналогичный результат может быть достигнут путем удаления из системы продуктов реакции. Знание принципов химического равновесия, основанных на правиле ЛеШателье, имеет очень большое практическое значение, поскольку дает возможность контроля химических реакций как в лаборатории, так и в промышленности. Необходимо отметить, что данный принцип применим только к системам, находящимся в состоянии истинного химического равновесия. Катализ Скорость химических реакций может возрастать не только при увеличении концентрации реагирующих веществ или температуры системы, но и под влиянием катализаторов. Вещества, которые увеличивают скорость химической реакции, оставаясь в конечном итоге неизменными по химическому составу и количеству, называют катализаторами. Процесс увеличения скорости реакции с помощью катализатора называется катализом, а реакции, в которых скорость изменяется в результате введения в реакционную смесь катализаторов, называются каталитическими. Особенности катализаторов: · Ускоряют реакцию, присутствуя в очень малых количествах · Избирательность действия, то есть катализатор ускоряет одну реакцию и неэффективен для другой. Особенно это свойство проявляется у биологических катализаторов-ферментов · Неизменность после реакции и возможность многократного использования · Катализатор изменяет механизм реакции и направляет ее по такому пути, который характеризуется понижением энергии активации. В зависимости от тог
|
|||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-01; просмотров: 452; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.138.34.93 (0.013 с.) |

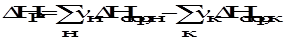
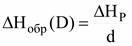
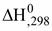 или
или  ) (стандартные теплоты образования).
) (стандартные теплоты образования).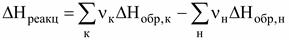
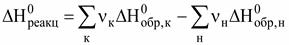

 ,
, .
.