
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Виды анализа (качественный, количественный, структурный). Анализ изотопный, молекулярный, функциональный, фазовый. Методы анализа (физ, хим и физ-хим)Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Виды анализа (качественный, количественный, структурный). анализ изотопный, молекулярный, функциональный, фазовый. Методы анализа (физ, хим и физ-хим) 1)Классиф-ия анализа в зависимости от цели: Качественный анализ –идентификация компонентов анализируемого в-ва. Количественный-определение конц. Или масс компонентов. Структурный-установление хим. И пространственного строения исслед.в-ва 2)класс-ция в зависимости от того,какие компоненты необходимо обнаружить: Изотопный(отдельные изотопы) Молекулярный(индивидуальное хим. Соед-ие, харак-ся определённой молекулярной Массой) Функциональный(функциональные группы) Фазовый(отдельные фазы в неоднородном объекте) Химические методы основаны на исп.хим.р-й(гравиметрия- определение масс малорастворимых осадков или газообразных продуктов р-ии,титриметрия-опред.к-во в-ва реагента,взаим. С определяемым в-вом) Физические методы анализа –измерение с пом.прибров физ.св-в определяемых в-в: эти св-ва изм-ся при изменении содержания в-ва в анализируемом объекте.Хим.р-ции не исп.К физ.методам относ.-спектроскопич.,электрометр.,термометрич.,радиометр. Физико-хим. -сочетают черты и физ. и хим. Измеряют физ. св-ва в-ва изм-ся в процессе протекания хим. р-ии.(молекулярно-абсорбционная спектроскопия-метод,осованный на измерении оптической плотности р-ра в-ва-физ. метод.Но если оптическая плотность измен-ся в процессе хим. р-ии,то метод становится физ-хим.)
Важанейшие хар-ки аналитической р-ии. Системат. метод анализа. Дробный метод анализа Предел обнаружения показывает какое минимальное количество определяемого вещества можно обнаружить с помощью данной методики. Избирательность показывает число в-в, вступающих в данную р-ию или взаимодействующих с данным реагентом. В зависимости от избирательности аналитические р-ии бывают: специфическими (обнаруживают 1 в-во), избирательными (позволяет обнаружить при данных условиях небольшое число в-в),групповыми(для выделения нек-ой группы в-в). Систематическим называется метод качественного анализа, основанный на разделении смеси ионов с помощью групповых реагентов на группы и подгруппы и последующем обнаружении ионов в пределах этих подгрупп с помощью селективных реакций. Название систематических методов определяется применяемыми групповыми реагентами. Известны систематические методы анализа: (сероводородный, кислотно-основный, аммиачно-фосфатный). Каждый систематический метод анализа имеет свою групповую аналитическую классификацию. Недостатком всех систематических методов анализа является необходимость проведения большого числа операций, длительность, громоздкость, значительные потери обнаруживаемых ионов и т.д. Дробным называется метод качественного анализа, предполагающий обнаружение каждого иона в присутствии других с использованием специфических реакций либо проведение реакций в условиях, исключающих влияние других ионов.Обычно обнаружение ионов дробным методом проводят по следующей схеме – вначале устраняют влияние мешающих ионов, затем обнаруживают искомый ион с помощью селективной реакции Способы устранения мешающего действия инов: - маскирование. исп. хим.р-ии,приводящие к уменьш. конц. мешающего ионами протекающие в той же фазе,что и основная р-ция.Для маскирования исп.р-ии комплексообразования и ОВР либо создают опреелённые значения рН.(при обнаруж. Ионов хрома(3) в виде дихромат ионов мешающие ионы железа(3) переводят в бесцветный фосфатный комплекс. - разделение. мешающие ионы удаляются из фазы, в кот.протекает р-ция.Разделение ионов м.б. проведено при пом. осаждения,экстракции, хроматографии.Напр.,Обнаружение ионов висмута(3) в присутствии ионов Со(2) в водном р-ре посредством р-ии с тиоцианат-ионами невозможно,т.к. комплекс(Со(SCN)4)2--синий,а (Вi(SCN)4—жёлтый.Изоамиловый спирт экстрагирует тиоционатный комплекс кобальта, а комплекс висмута ост. в р-ре.
Методы расчета концентрации вещества по величине аналитического сигнала: метод калибровочного графика, метод стандартов, метод добавок. Метод градуировочного графика: 1. Готовим серию стандартных р-ров с разными концентрациями, которые точно известны 2. Для каждого р-ра в одинаковых условиях получают величину аналитического сигнала (иногда используют внутренние стандарты – их содержание во всех пробах и в анализируемой одинаковое, тогда в качестве аналитического сигнала используем отношение сигналов определяемого в-ва к внутреннему стандарту) 3. Строим график (например, линейная зависимость у=а+bх) 4. Уравнения, описывающие градуировочный график, можно получить методом наименьших квадратов: коэффициенты a и b должны быть такими, чтобы сумма квадратов отклонений реальных значений от рассчитанных по полученному уравнению была бы минимальной. Метод стандартов: 1. Измеряем величину анал. сигнала для р-ра с известной концентрацией вещества 2. Для р-ра с неизвестной концентрацией 3. А) если уравнение не содержит свободного члена, то С = Сст Ух / Уст Б) если есть свободный член, то используем метод двух стандартов (одна чуть меньше, другая чуть больше) Сх = (С2 (Ух – У1) +С1 (У2 – Ух)) / (У2 – У1) Метод добавок (расчетный метод добавок) 1. Измеряем величину анал. сигнала для пробы с неизвестной концентрацией 2. Добавляем известное количество в-ва, измеряем новый сигнал 3. Сх = Сдоб Ух / (Удоб – Ух)
Качественный анализ катионов. Способы устранения мешающего влияния ионов. Устранение мешающего влияния катионов: 1. Маскирование (химические реакции, которые уменьшают концентрацию мешающих ионов, протекают в той же фазе, что и основная реакция): комплекс, протолитические, ОВР. Примеры: Co2+, Fe3+. Мешает Fe, его в комплекс [FeF6]-. Co2+ в осадок с SCN Протолитические р-ции – изменение рН среды. ОВР 2. Разделение (мешающие ионы удаляют из фазы, в которой протекает реакция): осаждение, экстракция, хроматография. Согласно кислотно-основной классификации катионы в зависимости от их отношения к растворам HCl, H2SO4, NaOH (или KOH) и NH3 разделяют на 6 групп. Каждая из групп, за исключением первой, имеет свой групповой реагент.(см далее) каждый отдельный катион может быть идентифицирован с помощью дробных реакций. Общая х-ка и аналитические р-ии катионов 1 аналит. группы. В 1 аналит. Группе относ-ся- K, Na, Li,NH4.Группового реагента нет,т.к. их сульфиды,гидроксиды,карбонаты,хлориды растворимы в воде.В ОВР не участвуют, за исключением NH4(облад. восстанов. св-вами).Не явл. комлексообразователями. Окрашивают пламя в жёлтый- Na,карминно-красный- Li,фиолетовый- К Li: Белый карбонат, белый фосфат, белый фторид. (+реакция с гексагидроксоантимонатом и цинк-уранил-ацетатом, хотя они на натрий) K: KCl + NaHC4H4O6 → KHC4H4O6 ↓ + NaCl (белый осадок) 2KCl + Na3[Co(NO2)6]→ 2NaCl + K2 Na[Co(NO2)6]↓(ярко-жёлтый) Na2Pb[Cu(NO2)6] + 2 KCl → К2Pb[Cu(NO2)6]↓ + 2 NaCl(кубические кристаллы чер- ного или коричневого цвета) Na: Na+ + [Sb(OH)6]- = Na[Sb(OH)6] мелкокристаллический белый(гексагидрокроантимонат) Натриевая соль метоксифенилуксусной кислоты (белый) - 2) Микрокристаллоскопическая реакция. на предметное стекло каплю раствора соли натрия-выпарить-Охлажденный осадок обработайте уранилацетатом UO2(CH3COO)2---образуются кристаллы NaCH3COO⋅ UO2(CH3COO)2. (игольчатые) NH4: NH4Cl + NaOH → NH3↑ + NaCl + H2O(запах аммиака) NH4++ 2[ HgI4]2 + 4OH-→ [OHg2NH2]I↓ + 7I + 3H2O(красно-бурый осадок)
Ртуть (II) Hg2+ + OH- = HgO↓ + H2O желтый Hg2+ + 2I- = HgI2↓ (оранжево-красный), при изб. Растворимый комплекс [HgI4]2- Hg2+ + Cu↓ = Hg↓ + Cu2+ (серебристый налет металлической ртути)
13. Систематический ход анализа катионов I-III аналитических групп катионов по кислотно-основной классификации. В исследуемом растворе могут содержаться ионы K, NH4, Pb, Ag, Ca, Ba 1. Предварительные испытания А) окраска и рН р-ра Б) +NaOH и индикаторная бумага (обнаруживаем ионы аммония) В) +HCl, если нет осадка, значит, нет Pb и Ag Г) +H2SO4, если нет осадка, нет Ca, Ba. !!!Pb дает осадки в в) и г), но его сульфат, в отличие от Са и Ва, растворим в NaOH Д) если нашли аммоний, то его удаляем прокаливанием, потом определяем 1 группу. +Na2Pb[Cu(NO2)6] (черные кубические кристаллы с калием) 2. 0.5 мл р-ра + 0.5 мл 2M HCl. Получили осадок, отфильтровали. В фильтрате обнаруживаем Ca, Ba. Осадок: KI на Pb, K2CrO4 на Ag Фильтрат: + серная, упаривают. Если игольчатые кристаллы, то это CaSO4 * 2H2O Для обнаружения Ba K2Cr2O7 в ацетатном буфере (желтый осадок, рН5, хроматы стронция и кальция в таких условиях не выпадают)
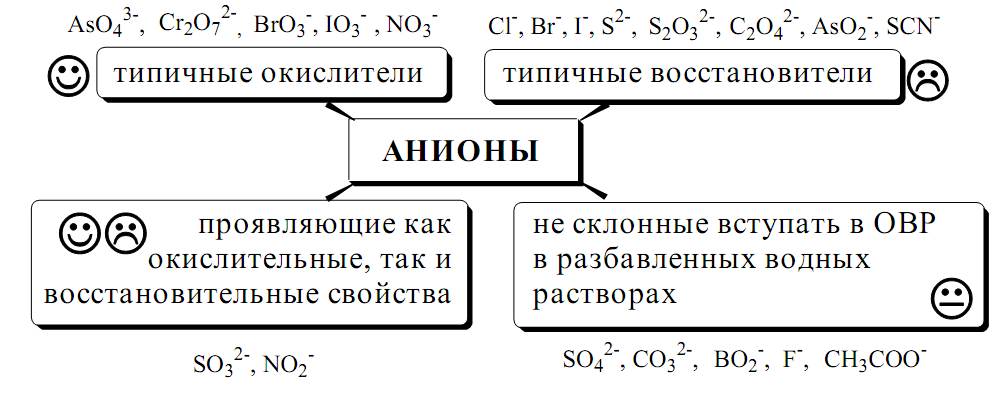
Аналитическая классификация аниоинов, групповые реагенты. Групповые реагенты в классификации ионов по растворимости солей бария и серебра, по окислительно-восстановительным свойствам. Предварительные испытания при анализе анионов. Реакции обнаружения анионов могут быть основаны на их окислительно-восстановительных свойствах, способности образовывать малорастворимые соединения, а также на взаимодействии с кислотами с образованием газообразных продуктов. Классификации анионов не являются строго установленными. Например, в зависимости от растворимости солей бария и серебра анионы разделяют на:
По окислительно-восстановительным свойствам анионы можно разделить на следующие группы:
Обнаружение анионов целесообразно начинать с предварительных испытаний: • установление рН раствора. Если среда кислая (рН<2), в ней не могут присутствовать анионы летучих и неустойчивых кислот (SO32¯, S2O32¯, CO32¯, NO2¯). Кроме того, в кислой среде в растворе не могут одновременно присутствовать анионы-окислители и анионы восстановители. • испытание на выделение газообразных веществ под действием разбавленных кислот. Исследуемый раствор обрабатывают 1 М H2SO4. Выделение СО2 указывает на присутствие СО32¯, SО2 на SО32¯, NО2 на NО2 ¯, одновременно SО2 и осадка S на присутствие S2О32-. Выделение I2 говорит об одновременном присутствии I ¯и анионов-окислителей. • испытание на присутствие анионов I группы. К исследуемому раствору добавляют раствор BaCl2 при рН 7-9. Отсутствие осадка S2О32¯ указывает на отсутствие анионов 1 группы, хотя BO2 образуют осадки с BaCl2 в концентрированных растворах. • испытание на присутствие анионов II группы. К исследуемому раствору добавляют раствор AgNO3 в присутствии разбавленного раствора НNO3. Отсутствие осадка указывает на отсутствие анионов 2 группы. • испытание на присутствие анионов-окислителей. К исследуемому раствору добавляют раствор KI в присутствии разбавленной H2SO4. Если при этом не выделяется I2, то анионы-окислители отсутствуют. • испытание на присутствие анионов-восстановителей. К исследуемому раствору добавляют раствор KMnO4 в нейтральной среде и нагревают. Выпадение темно-бурого осадка указывает на присутствие анионов-восстановителей. Дополнительно можно проверить наличие сильных восстановителей по обесцвечиванию раствора I2. Далее проводят реакции обнаружения анионов, отсутствие которых не было доказано в предварительных испытаниях. Раствор, в котором проводят обнаружение, не должен содержать никаких катионов кроме K ⁺, Na ⁺,NH4 ⁺. Мешающие катионы удаляют путем кипячения с раствором Na2CO3 (готовят «содовую вытяжку»). Анализ неизвестного неорганического вещества. Предварительные испытания. Переведение анализируемого вещества в раствор. Проведение анализа. Анализ неизвестного вещества можно условно разделить на 3 этапа: I. Предварительные испытания II. Переведение анализируемого вещества в растворимое состояние III. Обнаружение катионов и анионов Твердый образец измельчают и делят на 4 части: первая часть используется для предварительных испытаний, вторая – для обнаружения катионов, третья – для обнаружения анионов, четвертая – для повторного исследования. Предварительные испытания - органолептическое исследование (определение цвета, запаха, формы кристаллов, реакция среды с помощью индикатора, летучесть) - окрашивание пламени горелки (на очищенную, промытую и тщательно высушенную нихромовую или платиновую проволоку помещают пробу и вносят в пламя, разные металлы окрашивают пламя в различные цвета: натрий-желтый, калий-фиолетовый, медь-зеленый и т.д.) - действие кислот (при действии на пробу разб.минеральных кислот могут выделяться различные газы СО2,SO2,H2S,NO2) - растворение пробы в воде и определение рН раствора (если проба полностью растворяется в воде, то после определения рН можно сделать выводы о составе образца: в кислом растворе не могут содержаться соли неустойчивых кислот, в щелочном – соли слабых оснований, кроме того по окраске полученного раствора можно судить о наличии в образце окрашенных ионов(например, кобальта, никеля, меди, хрома)) Проведение анализа I. Обнаружение катионов - сначала проводят обнаружение ионов аммония, калия, натрия, лития, железа, ртути, мышьяка.
- затем определяют наличие II и III групп при помощи групповых реагентов соляной и серной кислот.
- далее на исследуемый раствор действуют изб.гидрокида нария в присутствии перекиси, в осадок выпадают катионы V и VI, а в оставшемся растворе определяют катионы IV группы.
- осадок V и VI групп обрабатывают раствором аммиака, в полученном растворе открывают катионы VI группы, а осадок исследуют на наличие катионов V группы.
II. Обнаружение анионов -мешающие катионы удаляются из раствора (кипячение раствора с избытком карбоната натрия → «содовая вытяжка»). Если исследуемый раствор содержит только катионы первой группы, то осаждение катионов не проводится. -прежде, чем приступить к обнаружению анионов, необходимо нейтрализовать раствор до рН = 7 уксусной или соляной кислотой, при нейтрализации в осадок выпадает гидроксид алюминия и гидрокарбонат меди. - этот осадок отделяют, а в фильтрате обнаруживают анионы I-III групп.
17. Химическое равновесие. Константа химического равновесия. Виды констант химического равновесия, используемые в аналитической химии (термодинамические, реальные и условные концентрационные, общие, ступенчатые) Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем. А2 + В2 ⇄ 2AB Константа равновесия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями исходных веществ и продуктов в состоянии химического равновесия. Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции. Константа химического равновесия не зависит от механизма реакции. Для записи выражения химического равновесия, необходимо знать лишь стехиометрическое уравнение реакции. В аналитической химии используют: 1. Термодинамические константы химического равновесия (К0 ) – выражают через активность частиц, принимающих участие в равновесии, которое характеризует соответствующая константа. Для равновесия aA+ bB=cC+dD, термодинамическая константа равновесия описывается: 2. Реальные концентрационные константы хим. равновесия (К)- предполагают использование равновесных концентраций частиц. Для ее расчета используют уравнение
3. Общие константы – сумма равновесных концентраций всех форм существования данного вещества. Отношение равновесной концентрации определенной формы вещества к общей концентрации этого вещества называется молярной долей данной формы вещества 4. Условные концентрационные константы хим. равновесия. Отличаются от реальных тем, что в описывающие их выражения входят общие концентрации веществ, участвующих в равновесии: 5. Равновесные константы – концентрация определенной формы вещества, участвующего в равновесии. Она представляет собой сумму равновесных концентраций всех форм существования данного вещества 6. Ступенчатые константы – константы, характеризующие каждую ступень. Произведение ступенчатых констант называется общей константой равновесия.
Существуют также смешанные констант равновесия, в которых, например, содержание одного иона выражено через активность, а остальных – через концентрации. В аналит. химии спользуются несколько типов химического равновесия, отличающихся друг от друга тем, обмен какими частицами происходит в процессе реакции. Константы равновесий могут быть очень малые или очень большие величины. Часто вместо значений констант равновесия более удобным оказывается использовать десятичные логарифмы. Отрицательный десятичный логарифм константы равновесия называется показателем данной константы и обозначается как рК. 18. Отклонения от идеальности в растворах сильных электролитов. Активность и коэффициент активности. Ионная сила раствора. Зависимость активности сильного электролита от ионной силы раствора. Активность(а) - это такая концентрация вещества в растворе, при использовании которой свойства данного раствора могут быть описаны теми же уравнениями, что и свойства идеального раствора. Активность называют действующей концентрацией. Измеряется моль/л. Активность может быть меньше номинальной концентрации, а может быть и больше. Активность чистого растворителя, нерастворенного вещества или жидкости, не смешивающейся с данным раствором, принимается равной 1. Отношение активности вещества в данном растворе к его концентрации называется коэффициентом активности. В зависимости от способа описания количественного состава раствора различают молярный(У), моляльный и рациональный (f) коэффициенты активности. Молярный коэффициент активности – отношение эффективной молярной концентрации веществ к его действительной молярной концентрации, моляльный – отношение эффективной моляльности к действительной, рациональный – отношение эффективной молярной доли к действительной молярной доле вещества в растворе. Заметное отклонение от идеальности имеется в растворах сильных электролитов. Это отражается на их температурах кипения и замерзания, давления пара над раствором, на величины различных констант равновесий в таких растворах. Для характеристики активности электролитов используются среднеионные коэффициенты активности, характеризующие поведение электролита в целом, и индивидуальные коэффициенты активности ионов: они характеризуют активность отдельных ионов, входящих в состав электролита. Активность вещества, находящегося в растворе электролита, зависит от концентрации всех ионов, присутствующих в нем и их заряда. Величина, которая учитывает влияние концентрации и заряда всех ионов, находящихся в растворе, на активность растворенного вещества, называется ионной силой: Ионная сила имеет размерность концентрации. При ее расчете учитывается концентрация и заряд всех ионов, образовавшихся при ионизации всех сильных электролитов, присутствующих в растворе. Неэлектролиты не оказывают влияния на величину ионной силы. Универсального уравнения, с помощью которого можно было бы рассчитать коэффициент активности любого электролита при любой величине ионной силы, не существует. Для описания зависимости коэффициента активности от ионной силы в очень разбавленных растворов (до ионной силы меньше О, О1) используют предельный закон Дебая-Хюккеля: При расчетах коэффициентов активности при ионных силах порядка 0,01- 0,1 используют расширенное уравнение Дебая-Хюккеля: При высоких значениях ионной силы(до 1) используют уравнение Дэвиса
Важнейшие теории кислот и оснований: протолитическая теория Бренстеда-Лоури, теория Аррениуса, теория Льюиса. Количественное описание силы кислот и оснований (константа кислотности, константа основности, константа кислотности сопряженной кислоты, их показатели). Теория Бренстеда-Лоури(протолитческая теория): кислоты - это вещества, способные отдавать протон(доноры протона), основания – это в-ва, способные присоединять протон(акцепторы протона). Кислоты и основания, согласно теории Бренстеда, существуют как сопряженные пары. Частица, содержащая на один протон больше, чем исходное основание, называется кислотой, сопряженный с данным основанием, а частица, содержащая на один протон меньше, чем исходная кислота, называется основанием, сопряженная с данной кислотой. Понятие «соль» в протолитической теории не используется. Протон, согласно этой теории, не существует в растворе в свободном виде, а связывается с молекулами растворителя. Теория Аррениуса: кислоты – это электролиты, при диссоциации которых в водном растворе образуются катионы только одного вида – катионы водорода, а основания – электролиты, при диссоциации которых в водном растворе в качестве анионов образуются только гидроксид-ионы. Кислота и основание могут взаимодействовать друг с другом с образованием соли и воды (р-ция нейтрализации). Если хотя бы один из компонентов, вступающих в реакцию нейтрализации, не относится к сильным электролитам, то данная реакция является обратимой. Реакция, обратная реакции нейтрализации – р-ция гидролиза. Применение теории Аррениуса, было ограничено только водными растворами, и далеко не все соединения подходили под определения «кислота» и «основание». Теория Льюиса (электронная теория кислот и оснований): основания – доноры пары электронов(кислоты, катионы металлов), кислоты – акцепторы пары электронов. Следствие Льюиса: любое органическое соединение можно представить как кислотно -основный комплекс. Теории Бренстеда и Льюиса являются наиболее широко использующимися теориями кислот и оснований в современной химии. Для количественной силы кислот, находящихся в растворе, используют константу, характериз. способность кислоты отдавать протон молекуле растворителя, выступающей в качестве основания. Такая константа называется константой кислотности (Ка). Константа кислотности характеризует равновесие и описывается выражениями Активность растворителя не входит в выражение для этой константы, т.к.считается равной 1. Термодинамическая константа кислотности для водных растворов описывается уравнением Силу оснований можно описывать двояко: либо с помощью константы основности(Кb), характеризующей равновесие Отрицательный десятичный логарифм константы кислотности сопряженной кислоты называется показателем константы кислотности сопряженной кислоты(рКвн+): рКвн+=-lgКвн+ Чем больше Квн+ и меньше рКвн+, тем сильнее сопряженная с описываемым основанием кислота и тем слабее само основание. 20. Классификация растворителей по кислотно-основным свойствам и полярности. Автопротолиз растворителя. Константа автопротолиза. Нивелирующее и дифференцирующее действие растворителя. Сильные и слабые кислоты и основания. Водородный показатель. Сила кислоты зависит от природы взаимодействующего с ней основания, а сила основания – от природы взаимодействующей с ним кислоты. В зависимости от наличия или отсутствия склонных к ионизации атомов водорода растворители можно условно разделить на протонные и апротонные. В составе молекул протонных растворителей имеется склонный к ионизации атом водорода (вода, спирты, карбоновые кислоты и т.д.). У апротонных растворителей подобных атомов водорода нет. В зависимотсти от диэлектрической проницаемости апротонные растворители можно разделить на 2 группы: неполярные апротонных растворители (гексан, бензол ит.д.) и полярные растворители (ацетон). В зависимости от кислотно-основных свойств можно выделить 4 группы растворителей: кислотные(протогенные), основные(протофильные), амфотерные и инертные. К группе кислотных растворителей относят в-ва, у которых преобладает кислотные свойства (уксусная кислота). Кислотные растворители повышают – по сравнению с водой – силу растворенных в них оснований и понижают силу кислот. У основных растворителей сильнее выражены основные свойства (аммиак) Основные растворители повышают силу растворенных в них кислот и понижают силу оснований. У амфотерных растворителей кислотные и основные свойства выражены приблизительно одинаково (спирты, вода). Инертные растворители практически не способны принимать участие в кислотно-основном взаимодействии(бензол). Автопротолиз – процесс кислотно-основного взаимодействия между двумя молекулами вещества, при котором одна молекула ведет себя как кислота, а вторая – как основание.Автопротолизу подвержено большинство растворителей. В реакции кислотно-основного взаимодействия между 2 молекулами растворителя: SH+SH=SH2++ S-. Частица SH2+ называется ионом лиония, а частица S- - ионом лиата. Равновесие можно охарактеризовать с помощью константы равновесия ( Отрицательный десятичный лагорифм константы автопротолиза называется показателем константы автопротолиза (рКSH). Константа автопротолиза является мерой протяженности шкалы кислотности. Кислоты, более сильные, чем ион лиония, или основания, более сильные, чем ион лиата, уравниваются по силе. Такое явление называется нивелирующим действием растворителя. Способность растворителя оказывать нивелирующее или дифференцирующее действие зависит от его кислотно-основных свойств и склонности к автопротолизу. Растворитель с сильными основными свойствами нивелирует силу кислот и дифференцирует силу оснований. Сильно кислотный растворитель, напротив, дифференцирует силу кислот и нивелирует силу оснований. Чем меньше величина константы автопротолиза растворителя, тем выше вероятность того, что он будет оказывать дифференцирующее действие на силу кислот и оснований. Водородный показатель(рН) – отрицательный десятичный логарифм активности ионов водорода в растворе: рН=-lg(H+). Шеала кислотности зависит от величины рКSH растворителя. Для характеристики кислотности и щелочности водных растворов используется интервал рН от 0 до 14. Количественная оценка окислительно-восстановительной способности веществ. Электродные потенциалы. Стандартный электродный потенциал полуреакции. Уравнение Нернста. Константа равновесия окислительно-восстановительной реакции. Для количественной оценки способности в-в отдавать и принимать электроны исп. электродные потенциалы. Электродный потенциал- разность потенциалов, кот. возникли на границе «металл-р-р» в рез-те разделения зарядов. Стандартный электродный потенциал полур-ции- это ЭДС гальванического элемента, сост. из нах. в станд. условиях электрода, на кот. протекает данная полур-ция, и стандартного водородного электрода. Гальвани́ческий элеме́нт — химический источник электрического тока, основанный на взаимодействии двух металлов и (или) их оксидов в электролите, приводящем к возникновению в замкнутой цепи электрического тока.
Стандартный водородный электрод пр.собой платиновую пластину, насыщенную водородом, кот.нах.в р-ре серной к-ты с а(Н+)=1. Уравнение Нернста: Уравнение Нернста описывает влияние активности компонентов, участв.в процессе, и температуры на величину потенциала. Взаимосвязь между общей концентрацией в-ва в р-ре и активностью его формы:
Константа равновесия окисл.-восст. р-ции:
ОВР широко исп.в титриметрии, для обнаружения неорг. и орг. в-в,для маскировки мешающих ионов, во многих электрохим.и кинетических методах анализа. С ним связаны процессы обмена веществ, протекающие в живом организме, гниение и брожение, фотосинтез. ОВР сопровождают круговорот веществ в природе. Их можно наблюдать при сгорании топлива, в процессах коррозии металлов, при электролизе и выплавке металлов другие ценные продукты. ОВР лежат в основе преобразования энергии взаимодействующих химических веществ в электрическую энергию в гальванических и топливных элементах. Температура Температура влияет как на константу равновесия ОВР, так и на их скорость. Как правило, ОВР обладают большим тепловым эффектом, поэтому изменение температуры оказывает значительное влияние на константу равновесия. Многие ОВР идут при комнатной температуре медленно (например, реакция окисления оксалат-ионов перманганат-ионами), и для их проведения требуется нагревание. Иногда, наоборот, нагревание является нежелательным. Посторонние ионы Присутствие посторонних индифферентных ионов в растворе приводит: 1. к повышению ионной силы раствора. Если коэффициенты активности окисленной и восстановленной формы при этом изменяются неодинаково, то изменяется и величина ОВ потенциала. 2. посторонние ионы могут оказывать влияние на скорость реакции. Анионы влияют на реакцию между катионами, катионы - на реакцию между анионами.
Влияние рН Ионы H+ могут: 1. сами участвовать в ОВР 2.окисленная или восстановленная форма может протонироваться, образуя новые ОВ пары.
Образование малорастворимых соединений Приводит к уменьшению концентрации окисленной или восстановленной формы и, сле- довательно, к изменению величины электродного потенциала.
Комплексообразование Окисленная или восстановленная форма либо они обе вместе могут связываться в комплексные соединения с ионами, присутст- вующими в растворе. Это приводит к изменению величины электродного потенциала. Отбор пробы газов Генеральная проба газообразных веществ небольшая. Используют вакуумные мерные колбы или бюретки с соответствующей запорной жидкостью, а также специальные контейнеры. При отборе пробы газов в замкнутом пространстве (например, в цеху, лаборатории) пробу отбирают в разных точках, а затем смешивают либо анализируют каждую из них отдельно.
Отбор пробы жидкостей Отбор пробы гомогенной жидкости (например, глазные капли или раствор для инъекций) проводят обычно по объёму, используя для этой цели пипетки или бюретки. Предварительно жидкость тщательно перемешивают. Если анализируемую жидкость сложно или невозможно перемешать,то отбор пробы проводят на разной глубине ёмкости. Гетерогенные жидкости перед взятием пробы тщательно гомогенизируют путём перемешивания либо вибрации. Пробы таких жидкостей часто отбирают не только по объёму, но и по массе. Если анализируют жидкость из потока, то для получения достоверной информации пробы отбирают из различных мест по течению водотоков.
Отбор проб твёрдых веществ Величина генеральной пробы твёрдого вещества зависит от неоднородности образца и размера частиц. Оценка массы генеральной пробы- формула Ричердса-Чеччота Масса пробы должна быть такой, чтобы погрешность, обусловленная отбором пробы, не превышала 4/5 общей погрешности результата анализа. Целый твёрдый объект перед отбором из него пробы измельчают Объекты, представляющие собой сыпучие вещества, перемешивают и затем берут пробу из разных частей.
Причины погрешностей при отборе проб: • потерями компонентов в виде пыли; • потерями летучих веществ; • взаимодействием компонентов пробы с кислородом воздуха, материалом посуды; • адсорбцией компонентов пробы на поверхности посуды.
Разложением пробы называют процесс переведения определяемых компонентов пробы в физическую и химическую форму, которая наиболее приемлема для выбранного метода определения. Термическое разложение Термическое разложение пробы проводят путём её нагревания до высокой температуры (иначе говоря, путём сжигания пробы) на воздухе или в атмосфере кислорода. Термическое разложение пробы чаще всего проводят путём прокаливания её на воздухе в открытых чашках и тиглях при температуре 500-600 °С или сжиганием в колбе, заполненной кислородом. Прокаливание на воздухе в открытых сосудах используется для определения зольности органических веществ, при определении тяжёлых металлов в биологических объектах (один из способов «сухой» минерализации). К такому способу разложения пробы прибегают тогда, когда объектом для последующего анализа должно быть твёрдое вещество, а не раствор (например, если анализ будет проводиться атомно-эмиссионным или ре
|
||||
|
|
Последнее изменение этой страницы: 2016-06-29; просмотров: 1081; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.22.217.176 (0.013 с.) |


 .
. Схема гальванического элемента Даниэля-Якоби.
Схема гальванического элемента Даниэля-Якоби.
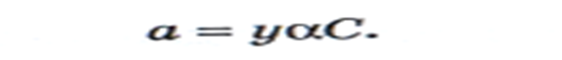
 Взаимосвязь константы равновесия ОВР и ЭДС:
Взаимосвязь константы равновесия ОВР и ЭДС:



