
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Методы контроля качества основных (базисных) гомеопатических препаратовСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Контроль качества основных гомеопатических препаратов можно подразделить на 2 этапа: а) контроль физико-химических свойств и технологических параметров; б) аналитический контроль по действующим веществам. Жидкие базисные препараты (эссенции, настойки, растворы) контролируют в соответствии с требованиями руководства В. Швабе «Гомеопатические лекарственные средства» и ГФ по следующим показателям: Ø соответствие запаха и вкуса; Ø прозрачность (отсутствие механических включений); Ø соответствие окраски, т. к. ряд препаратов, особенно приготовленных из свежих растений при длительном хранении изменяют свою окраску (например, часто наблюдается изменение зеленой окраски в коричневую, вызванное в большинстве случаев изменением хлорофилла). Кроме того, может также изменяться интенсивность окраски в различных пробах одного и того же препарата, несмотря на равное содержание лекарственного вещества, что особенно заметно в самых высоких разведениях. Этот факт необходимо учитывать при оценке приведенных сведений об окраске различных веществ. Окраску определяют визуально при дневном отраженном свете на матово-белом фоне (белый картон или писчая бумага) в пробирках одинакового стекла диаметром 10 мм. Ø капиллярный и капиллярно-люминесцентный анализ: а) капиллярный анализ эссенций, настоек и жидких разведений проводят по методу «Плана»: из фильтровальной или хроматографической бумаги одного сорта в направлении, перпендикулярном текстуре бумаги, нарезают полоски шириной 2 см и длиной приблизительно 25 см и подвешивают в цилиндрическом стеклянном сосуде, высотой около 5 см и диаметром около 3 см так, чтобы концы бумажных полосок касались дна сосудов. В сосуд, если не оговорены другие условия проведения анализа, помещают обычно 5 мл исследуемого раствора. Сосуд ставят в умеренно теплое помещение и через 24 часа или к моменту, когда вся жидкость будет поглощена вынимают полоски, просушивают и исследуют при дневном свете или же в ультрафиолетовом свете, излучаемом кварцевой аналитической лампой. При исследовании более высоких разведений вместо широких капиллярных полосок используются полоски шириной не более 2,5 мм. При описании капиллярной картины пользуются делением на 2 части: Верхняя часть, состоящая из водной зоны и часто зоны в виде выпуклости или эллиптической выемки. Нижняя часть, большей частью состоящая из нескольких зон, окрашенных в разные цвета, и основания. Контролем служат данные капиллярного анализа эссенций или настоек, приведенные для каждого объекта в руководстве В.Швабе; б) капиллярно-люминесцентный анализ, разработанный Нейгебауэром и Платцем, принятый в международной гомеопатической фармакопее, уточнен и приспособлен для условий аптеки или лаборатории как метод, дающий ясную картину специфичности средства и правильности приготовления лекарств. При наблюдении люминесценции жидкости исследуемой методом капиллярного анализа целесообразнее всего также оказалось разделение на 2 части: Верхнюю часть, которая состоит из узкой самой верхней зоны, затем собственно верхней части и основания верхней части, ясно наблюдаемого при люминесценции целого ряда препаратов. Нижнюю часть, которая состоит из выпуклой части или свода, полосы, состоящей из нескольких зон и основания; полоса может занимать всю нижнюю часть или только выпуклую зону. Данные капиллярного анализа наблюдают при свете аналитической УФ-лампы обычно после просушки, так как при этом наиболее полно проявляется характерная люминесценция. При наблюдении капиллярных картин в ультрафиолетовом свете для того, чтобы избежать ошибок, необходимо обращать внимание на следующее: как при дневном свете, так и при освещении лампой наблюдение нужно всегда проводить на одинаковом фоне, лучше всего белом, по возможности не люминесцентном. Кроме того, надо знать, что и от фильтровальной бумаги появляется, как правило, бледно-голубая или сине-фиолетовая люминесценция, а также, что различные вещества, как, например, молочный или тростниковый сахар, имеют часто собственную люминесценцию голубого цвета, которая может проявляться также при исследовании спиртового экстракта и затруднять определение вещества. Этиловый спирт также имеет слегка голубую люминесценцию. Нужно следить также за тем, чтобы у холостых проб с очищенной водой, на верхнем конце капиллярных картин всегда появлялась узкая зона, окрашенная в коричневатый цвет. В ультрафиолетовом излучении она светится ярко-синим светом. В целях более точного исследования препарат нужно обработать соответствующими реактивами, после чего можно наблюдать характерные изменения окраски при дневном, а особенно ультрафиолетовом свете. Рекомендуется обильно наносить раствор на все зоны стеклянной палочкой или капельной пипеткой и высушивание проводить при слегка повышенной температуре. В сомнительных случаях рекомендуется проводить холостую пробу на той же полоске бумаги, но выше капиллярной картины. Если применение реактивов недостаточно для доказательства идентичности, то можно использовать описанный ниже метод (2-я капилляризация): исследуемую фильтровальную бумагу с капиллярной картиной помещают в пробирку, затем наливают (до верхней границы капиллярной картины) соответствующий растворитель, чаще всего хлороформ (чтобы помешать тому, чтобы верхняя часть картины случайно не была бы барьером для растворителя и веществ, растворяющихся в нем). Растворитель растворяет все содержащиеся в окрашенном участке фильтровальной бумаги растворимые вещества и вместе с ними поднимается по бумаге. Затем эти вещества испаряются и отлагаются в новой зоне, на верхнем краю пробирки. Если эта «новая зона» получается слишком слабой, то опыт можно повторить с добавочной порцией растворителя, и следовательно повысить интенсивность этой зоны. Если эта зона слишком темна, то можно снова нанести растворитель, расширить этим зону и, таким образом, просветлить её. Новая более или менее широкая зона имеет часто характерный цвет и при дневном свете и при ультрафиолетовом освещении. В случае необходимости ее исследование, как и капиллярной картины, можно продолжать разными методами. Растворы проверяют на люминесценцию непосредственно; для этого 1-2 мл помещают в пробирку диаметром около 1,5 см и наблюдают в ультрафиолетовом свете. К раствору добавляют несколько капель хлористоводородной кислоты, чтобы исключить, особенно при высоких разведениях, помехи, вызываемые влиянием имеющейся щелочи; Ø определение плотности жидкостей проводят с помощью пикнометра или ареометра: Метод 1. Применяют в случае определения плотности жидкостей с точностью до 0,001. Чистый сухой пикнометр взвешивают с точностью до 0,0002 г, заполняют с помощью маленькой воронки очищенной водой немного выше метки, закрывают пробкой и выдерживают в течение 20 мин в термостате, в котором поддерживают постоянную температуру воды 20°С с точностью до 0,1°С. При этой температуре уровень воды в пикнометре доводят до метки, быстро отбирая излишек воды при помощи пипетки или свернутой в трубку полоски фильтровальной бумаги. Пикнометр снова закрывают пробкой и выдерживают в термостате еще 10 мин, проверяя положение мениска по отношению к метке. Затем пикнометр вынимают из термостата, фильтровальной бумагой вытирают внутреннюю поверхность горлышка пикнометра, оставляют под стеклом аналитических весов в течение 10 мин и взвешивают с той же точностью. Пикнометр освобождают от воды, высушивают, споласкивая последовательно спиртом и эфиром (сушить пикнометр путем нагревания не допускается), удаляют остатки эфира продуванием воздуха, заполняют пикнометр испытуемой жидкостью и затем производят те же операции, что и с очищенной водой. Плотность r20 (г/см3) вычисляют по формуле:
где m – масса пустого пикнометра в граммах; m1 – пикнометра с очищенной водой в граммах; m2 – масса пикнометра с испытуемой жидкостью в граммах; 0,99703 – значение плотности воды при 20°С (в г/см3 с учетом плотности жидкостей с учетом плотности воздуха); 0,0012 – плотность воздуха при 20°С и барометрическом давлении 1011 гПа (760 мм рт.ст.). Метод 2. Применяют в случае определения плотности жидкостей с точностью до 0,01. Испытуемую жидкость помещают в цилиндр при температуре жидкости 20°С осторожно опускают в нее чистый сухой ареометр, на шкале которого предусмотрена ожидаемая величина плотности. Ареометр не выпускают из рук до тех пор, пока не станет очевидным, что он плавает; при этом необходимо следить, чтобы ареометр не касался стенок и дна цилиндра. Отсчет производят через 3-4 мин после погружения по делению на шкале ареометра, соответствующему нижнему мениску жидкости (при отсчете глаз должен быть на уровне мениска). В случае определения темноокрашенных жидкостей отсчет производят по верхнему мениску. При точном соблюдении правил приготовления эссенции по описаниям +отдельных §§ плотность основных настоек в среднем равен: по §1 – 0,944 по §2 – 0,944 по §3 – 0,905 Ø определение содержания экстрактивных веществ (сухого остатка): выпаривают на водяной бане точно измеренное и точно взвешенное (с учетом плотности) количество жидкости, которое помещают в предварительно взвешенную фарфоровую чашку диаметром в 6-7 см. Затем сушат в течение 30 минут в термостате при 105°С. Взвешивать следует по возможности быстрее, так как некоторые экстракты очень сильно поглощают влагу и поэтому масса их увеличивается на весах в течение нескольких минут. Также не следует сушить долее получаса, так как при длительной сушке при 105° С масса жиросодержащих сухих остатков вновь возрастает. Содержание экстрактивных веществ (Х, %) рассчитывают по формуле:
m – масса навески препарата до высушивания, г; m1 – масса сухого остатка после высушивания, г; Ø определение содержания жирных растительных масел: остаток, получаемый при определении содержания экстрактивных веществ, смачивают 1-2 мл воды (иногда с подогревом на водяной бане), а затем растирают до получения однородного порошка с 10,0 г прокаленного гипса. Массу помещают в гильзу из фильтровальной бумаги и накрывают ватным тампоном. Гильзу помещают в аппарат Сокслета и экстрагируют в течение 2–3 часов слегка кипящим петролейным эфиром. Затем эфир отгоняют, остаток сушат в течение 15 минут в сушильном шкафу при температуре 105° и взвешивают; Ø количество обезжиренного сухого остатка определяют путём вычитания количества жирных масел из общего содержания сухого остатка. Ø определение содержания нерастворимого в воде осадка в экстрагируемом остатке настоек и эссенций, приготовленных по §§1–3: 25,0 г эссенции выпаривают на водяной бане и непродолжительное время сушат в сушильном шкафу при температуре 105°С. После охлаждения остаток разбавляют водой, растирают и фильтруют через точно взвешенный фильтр и промывают водой. Затем фильтр высушивают и взвешивают. Содержание нерастворимого осадка вычисляют по отношению к 100 частям экстрагируемого остатка настоек и эссенций. Ø определение содержания этилового спирта: а) по плотности отгона: в круглодонную колбу вместимостью 200–250 мл отмеривают точное количество жидкости (если жидкость содержит от 20 до 50% спирта – 50 мл, от 50% и выше – 25 мл; жидкость перед перегонкой разбавляют водой до 75 мл). Для равномерного кипения в колбу с жидкостью помещают капилляры, пемзу или кусочки прокаленного фарфора. Если жидкость при перегонке сильно пенится, то добавляют фосфорную или серную кислоту (2–3 мл), хлорид кальция, парафин или воск (2–3 г). Приёмник (мерную колбу вместимостью 50 мл) помещают в сосуд с холодной водой, собирают около 48 мл отгона, доводят его температуру до 20°С и добавляют воды до метки. Отгон должен быть прозрачным или слегка мутноватым. Плотность отгона определяют пикнометром и по алкоголеметрическим таблицам находят соответствующее содержание спирта в процентах по объему и массе. Содержание спирта в препарате (Х) в процентах по объёму вычисляют по формуле:
50 – объём отгона в миллилитрах; а – содержание спирта в процентах по объёму; б – объем исследуемого препарата, взятый для отгона, мл. При содержании в жидкости эфирных масел, летучих кислот или оснований, камфоры к ней добавляют в делительной воронке равный объем насыщенного раствора натрия хлорида и такой же объем петролейного эфира. Смесь взбалтывают в течение 3 минут. После разделения слоёв спиртоводный слой сливают в другую делительную воронку и обрабатывают таким же образом половинным количеством петролейного эфира. Спиртоводный слой сливают в колбу для отгона, а соединённые эфирные жидкости взбалтывают с половинным количеством насыщенного раствора натрия хлорида, потом присоединяют к жидкости, находящейся в колбе для отгона. При содержании летучих кислот их нейтрализуют раствором щелочи, при содержании летучих оснований – фосфорной или серной кислотой; б) по температуре кипения настоек: прибор для количественного определения спирта в настойках состоит из сосуда для кипячения 1, трубки 2 с боковым отростком, холодильника 3, ртутного термометра 4 с ценой деления 0,1°С и пределом шкалы от 50 до 100°С (см. рис.).
Полученный результат приводят к нормальному давлению. Если показания барометра отличаются от 1011 гПа (760 мм рт. ст.), вносят поправку на разность между наблюдаемым и нормальным давлением 0,04°С на 1,3 гПа (1мм рт. ст.). При давлении ниже 1011 гПа поправку прибавляют к установленной температуре, при давлении выше 1011 гПа поправку вычитают. Содержание спирта в настойке определяют при помощи табл. 18. Пример. Температура кипения настойки пустырника 80,9°С, атмосферное давление 1000 гПа (752 мм рт. ст.), разность давлений 1011 – 1000 = 11 гПа (760 – 752 = 8 мм рт. ст.). поправка составляет: 0,04°С × 8 = 0,32°С. К найденной температуре кипения прибавляют поправку: (80,9 + 0,32)°С. По табл. 18 этой температуре кипения соответствует 66% спирта. Таблица 18 Определение концентрации спирта в водно-спиртовых смесях
в) по показателю преломления жидкостей: в водных растворах этилового спирта линейная зависимость показателя преломления и концентрации наблюдается в пределах до 50-60%. При установлении крепости спирта в более концентрированных растворах следует их предварительно разбавить и при расчетах концентрации учитывать разведение.
Таблица 19 Показатели преломления спирто-водных растворов,
При определении показателя преломления спирто-водных растворов следует на призму рефрактометра наносить не менее 5-7 капель и измерять величину n немедленно во избежание ошибки, связанной с летучестью спирта. Исследование необходимо проводить при температуре 20°С. Если оно осуществляется не при 20°С, следует вносить поправки на температуру. Величины поправок показателя преломления на 1°С представлены в табл.19. Если определение проводится при температуре выше 20°С, то поправку прибавляют к найденной величине показателя преломления; если анализ проводится при температуре ниже 20°С, поправку вычитают. Пример. Анализу подвергался 40% спиртовой раствор. Определение показателя преломления проводили при 23°С. Показание рефрактометра – 1,3541. Согласно табл.19 поправка на 1°С для показателя преломления, близкого по величине к полученному (1,35500), равна 2,4×10-4 (т. е. 0,00024). Поскольку исследование проводилось при 23°С, то поправка будет составлять 0,00024 × 3 = 0,00072. Показатель преломления, приведенный к 20°С, равен 1,3541 + 0,00072 = 1,35482. По табл.19 определяют соответствующую данному показателю преломления концентрацию спирта. Найденной величины показателя преломления (1,35482) в таблице нет; близкому по величине показателю преломления 1,35500 соответствует 40% спирт. Необходимо определить, какая концентрация спирта соответствует разности показателей преломления: 1,35500 – 1,35482 = 0,00018. Поправка на 1% спирта равна 4,0×10-4. Следовательно, Для определения концентрации этилового спирта в спиртовых растворах лекарственных препаратов, приготовленных на 70% спирте, разбавление проводят обычно 1:2, а приготовленных на 90% и 95% спирте, – 1:3. При этом необходимо учитывать, что при смешивании спирта с водой объем раствора несколько уменьшается, в связи с чем следует вносить поправку к фактору разведения: при смешивании 1 мл спирта с 2 мл воды умножают коэффициент 2,98 (вместо 3); при смешивании 1 мл спирта с 3 мл воды – на 3,93 (вместо 4). Пример. Анализировали настойку барбариса, приготовленную по §4 на 70% спирте; определение проводили при 20°С. По показаниям рефрактометра n = 1,34555. В табл.12 данная величина показателя преломления отсутствует, наиболее близким значением является 1,34573, что соответствует 23% концентрации спирта. Разность показателей преломления составляет: 1,34573 – 1,34555 = 0,00018. Поправка на 1% спирта по табл.12 равна 6,1×10-4, следовательно, разности показаний соответствует концентрация спирта г) по плотности жидкости, определенной с помощью ареометра: по алкоголеметрическим таблицам ГФ находят соответствующее содержание спирта в процентах по массе и по объему; Ø определение содержания тяжелых металлов: в фарфоровой чашке упаривают досуха 5 мл жидкого исследуемого препарата, затем остаток осторожно сжигают в присутствии серной кислоты и прокаливают. Полученный остаток обрабатывают при нагревании 5 мл насыщенного раствора аммония ацетата, фильтруют через беззольный фильтр и доводят до метки 100 мл. 10 мл полученного раствора должны выдерживать испытания на тяжелые металлы (не более 0,001%). Для контроля качества порошковых растираний (тритураций) проводят следующие определения: Ø равномерность распределения лекарственных веществ в тритурациях: порошки рассматривают на расстоянии 20–25 см с помощью лупы или микроскопа с окулярным микрометром в прямом свете: лекарственное вещество должно быть равномерно распределено в молочном сахаре; Ø соответствие окраски, вкуса, запаха: в низких разведениях у окрашенных, сильно пахнущих и имеющих резкий вкус исходных веществ можно заметить соответствующую окраску и почувствовать своеобразный запах или вкус; Ø однородность: основная масса готовой тритурации должна состоять из частиц размером 25 мкм и менее, не должно быть частиц размером более Ø величина внешней удельной поверхности тритурации должна быть не менее 0,65 м2/г, а молочного сахара – не менее 0,50 м2/г; Ø размер частиц металлических и угольных растираний: на предметное стекло наносят 0,02-0,03 г соответствующего растирания, добавляют 1-2 капли воды и вызывают растворение молочного сахара умеренным нагреванием; затем (при не очень высокой температуре) раствор выпаривают настолько, чтобы остался вязкий, олифоподобный остаток, который накрывают покровным стеклом. Препарат рассматривают под микроскопом при увеличении в 200 раз, а величину непрозрачных металлических частичек определяют с помощью окулярного микрометра; Ø капиллярный анализ: растирания берут в количестве 5 г, смешивают примерно с двойным весовым количеством абсолютного этилового спирта и полученную смесь подвергают капиллярному анализу как жидкое разведение; Ø перекристаллизация насыщенных растворов: взвешенную пробу вещества помещают в мерную колбу с определенным количеством воды, различным для каждого вещества, а колбу покрывают небольшим кристаллизатором. Растворения достигают нагреванием закрытой колбы в кипящей воде или на открытом пламени, затем медленно охлаждают на воздухе: а) с веществами, пересыщенные растворы которых полностью кристаллизуются при соприкосновении с изоморфным кристаллом, поступают следующим образом: небольшой пипеткой осторожно берут несколько капель пересыщенного раствора и помещают по одной на стеклянную пластинку, затем небольшим, предварительно прокаленным, а затем полностью охлажденным платиновым шпателем берут небольшую пробу (приблизительно величиной с булавочную головку) растирания, подлежащего испытанию, и помещают ее в одну из капель пересыщенного раствора, находящегося на стеклянной пластинке. Если в пробе был хоть один изоморфный кристалл, то сравнительно быстро происходит кристаллизация всей капли, в результате чего образуется грубая кристаллическая поверхность и одновременно теряется ее прозрачность. Примером этого класса веществ являются натрия ацетат и сегнетова соль; б) вещества, пересыщенные растворы которых, соприкасаясь с изоморфным кристаллом, увеличивают его, а сами при этом не кристаллизуются: с помощью пипетки берут несколько миллилитров пересыщенного раствора и осторожно, так, чтобы не смочить край и верхнюю поверхность стенки, помещают в маленькую пробирку, закрываемую резиновой пробкой. С помощью маленького, предварительно прокаленного и полностью охлаждённого платинового шпателя, добавляют к раствору небольшую пробу исследуемого растирания, пробирку закрывают резиновой пробкой, осторожно опрокидывают и оставляют в наклонном положении на несколько часов. Если в пробе были микроскопические изоморфные кристаллы, то через несколько часов на нижней стенке можно заметить некоторое количество выросших кристаллов или друз различной величины. Примером этого класса веществ являются бура и меди сульфат. Примечание: у растираний веществ, которые в пересыщенных растворах могут вызывать явление перекристаллизации, этот метод можно использовать для проверки приготовления лекарства согласно прописи, т.к. явление перекристаллизации наблюдается и при высоких разбавлениях, например, таких как с 5 до 9 десятичные растирания. Аналитический контроль* качества основных гомеопатических препаратов по действующим веществам проводят различными методами в зависимости как от природы исходных веществ, так и от того являются ли они фармакопейными или не фармакопейными препаратами. Фармакопейные аллопатические препараты, которые применяются также в гомеопатии, анализируют на подлинность и количественное содержание по методикам фармакопейных изданий. В качестве примеров можно привести некоторые химические соединения, применяемые для приготовления растворов или порошковых растираний: меди сульфат (CuSO4 ·5H2O) подлинность: реакция с железом металлическим (проба на Cu2+); реакция с аммиаком (проба на Cu2+); реакция с бария нитратом (проба на SO42–); количественное определение: субстанция – йодометрическое определение; тритурации, дилюции – йодометрическое определение – до Х3; фотоколориметрически с аммиаком – до Х4; калия карбонат (K2CO3) подлинность: реакция с винной кислотой (проба на K+); реакция с хлороводородной кислотой (проба на CO32-); количественное определение: субстанция – кислотно-основное титрование; тритурации, дилюции до Х3 – кислотно-основное титрование; потенциометрическое титрование; прямая потенциометрия; натрия карбонат (Na2CO3) подлинность: реакция с цинкуранил ацетатом (проба на Na+); реакция с хлористоводородной кислотой (проба на CO32-); количественное определение: субстанция – кислотно-основное титрование; тритурации, дилюции до Х3 – кислотно-основное титрование; потенциометрическое титрование; прямая потенциометрия; бура, натрия тетраборат (Na2B4O7 ·10H2O) подлинность: реакция образования борноэтилового эфира; количественное определение: субстанция – кислотно-основное титрование; тритурации, дилюции до Х3 – кислотно-основное титрование; потенциометрическое титрование; прямая потенциометрия; натрия гидрокарбонат (NaHCO3) подлинность: реакция с цинкуранил ацетатом (проба на Na+); реакция с хлористоводородной кислотой (проба на CO32–); реакция с магния сульфатом (проба на HCO3–); количественное определение: субстанция – кислотно-основное титрование; тритурации, дилюции до Х3 – кислотно-основное титрование; потенциометрическое титрование; прямая потенциометрия; Натрия хлорид (NaCl) подлинность: реакция с цинкуранил ацетатом (проба на Na+); реакция с серебра нитратом (проба на Cl–); количественное определение: субстанция – аргентометрическое определение по Мору; тритурации, дилюции до Х3 – аргентометрическое титрование; потенциометрическое титрование; прямая потенциометрия; магния сульфат (MgSO4 ·7H2O) подлинность: реакция с натрия гидрофосфатом в аммиачном буфере (проба на Mg2+); реакция с бария нитратом (проба на SO42–); количественное определение: субстанция – комплексонометрическое титрование; тритурации, дилюции до Х3 – аналогично;
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-04-19; просмотров: 556; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.33 (0.011 с.) |

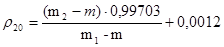 ,
,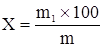 , где:
, где: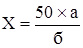 , где:
, где: В сосуд для кипячения наливают 40 мл настойки и для равномерного кипения помещают капилляры, пемзу или кусочки прокаленного фарфора. Термометр помещают в приборе таким образом, чтобы ртутный шарик выступал над уровнем жидкости на 2–3 мм.
В сосуд для кипячения наливают 40 мл настойки и для равномерного кипения помещают капилляры, пемзу или кусочки прокаленного фарфора. Термометр помещают в приборе таким образом, чтобы ртутный шарик выступал над уровнем жидкости на 2–3 мм.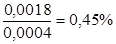 . Таким образом, истинное содержание спирта в исследуемом растворе 40 – 0,45 =39,55%.
. Таким образом, истинное содержание спирта в исследуемом растворе 40 – 0,45 =39,55%.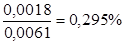 . Так как перед определением настойку разводили 1:2, истинная концентрация составляет (23 – 0,295) × 1,98 = 67,66%.
. Так как перед определением настойку разводили 1:2, истинная концентрация составляет (23 – 0,295) × 1,98 = 67,66%.


