
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Основні позначення та формули для обчислень у титриметріїСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
A, B, C,...F — інгредієнти (елемент, іон, сполука); X — інгредієнт, який визначають; R — реагент (робочий розчин); RН — реагент, доданий у надлишку; RТ — реагент, яким титрують; m — маса речовини, г; g — наважка зразка, г; V — об’єм розчину, мл; M — молярна маса, г/моль; r — густина розчину, г/см3; n — кількість молів речовини; w — масова частка інгредієнта; С — молярна концентрація (молярність), моль/л; СЕ — молярна концентрація еквівалента ( нормальність), моль/л; E — молярна маса еквівалента, г/моль; fE — фактор еквівалентності; T — титр розчину, мг/мл; T(R/X) — титр розчину реагенту за визначуваною речовиною. Перша група наведених формул (3.1) - (3.10) дає змогу обчислити кон-центрацію розчинів. У круглих дужках після відповідного позначення вказано інгредієнт, для якого це обчислення виконують: w(A) = C(A) =
CE(A) = fE(A) = T(A) = w(A) = C(A) = CE(A) = T(R/X) =
Формули (3.11) - (3.18) використовують для обчислень у титриметичному аналізі:
m(X) = w(X) = Масу інгредієнта X або його масову частку w(X) у випадку титрування робочим розчином (R) обчислюють за формулами: m(X) = w(X) = При титруванні залишку (обернене титрування) використовують такі формули для обчислень: m(X) = w(X) = Якщо визначення виконують методом піпетування, то масу m(X) та масову частку інгредієнта w(X) перераховують на весь початковий об’єм роз-чину, в якому розчинено наважку зразка. Для цього одержане значення збільшують у k разів, де число k вказує, у скільки разів об’єм мірної колби Vк більший від об’єму піпетки Vп k = 3.3. ПРОТОЛІТОМЕТРІЯ (КИСЛОТНО-ОСНОВНЕ ТИТРУВАННЯ ) До цього методу належать визначення, в основі яких є реакція H3O+ + OH- За допомогою цього методу можна визначати кислоти, луги, солі слабких кислот і основ. Прямим титруванням визначають слабкі протоліти, константа протолізу яких не дуже мала (К ³ 10-8). При титруванні поліпротонних протолі-тів на кривій титрування спостерігається декілька стрибків, якщо співвідно-шення констант Kт досить велике: Первинними стандартами в методі протолітометрії є розчин натрію тетраборату (Na2B4O7.10 H2O) для стандартизації розчинів сильних кислот та оксалатної кислоти (H2C2O4. 2 H2O) для стандартизації розчинів сильних основ. Вторинними стандартами є розчини сильних кислот (HCl, HNO3, H2SO4) та лугів (NaOH, KOH), тобто речовин, за наважкою яких не можна виготовити розчин точно відомої концентрації. Оскільки більшість розчинів, які використовують у протолітометрії, є безбарвними, то для фіксування точки стехіометричності використовують індикатори. Принцип дії індикаторів пояснює іонно-хромофорна теорія. Кожен з індикаторів має проміжок значень pH, у якому він змінює свій колір. Цей інтервал зміни забарвлення визначають в одиницях pH за формулою pH = pKHInd ± 1, (3.19) де KHInd — константа протолізу індикатора. Індикатор для титрування підбирають так, щоб його інтервал переходу збігався зі стрибком на кривій титрування. Можна підбирати його і так, щоб обчислене значення pH розчину в точці еквівалентності припадало на інтервал переходу індикатора. Приблизно по середині інтервалу переходу індикатора є значення pH, при якому він найрізкіше змінює свій колір: це показник титру-вання pT. Характеристики найбільш вживаних індикаторів наведені в табл. 3.1. Таблиця 3.1.Характеристики деяких індикаторів методу протолітометрії
Визначення основ Концентрацію розчину основи визначають за допомогою титрування її робочим розчином кислоти. Для стандартизації робочого розчину кислоти як первинний стандарт використовують натрій тетраборат, Na2B4O7. 10 H2O, який взаємодіє з кислотою за рівнянням: B4O72- + 2 H3O+ + 3 H2O Реагенти: Na2B4O7. 10 H2O, перекристалізований; HCl, концентрований розчин; NaOH, ~0,1 М розчин; Na2CO3, ~0,1 М розчин; індикатори: метиловий оранжевий, метиловий червоний. Приготування розчину натрію тетраборату. Натрій тетраборат при зберіганні втрачає частину кристалізаційної води, тому перед приготуванням цю сіль потрібно перекристалізовувати. Для титрування використовують 0,05 М розчин, який готують у мірній колбі на 200 або 250 мл. За формулою (3.12) обчислюють необхідну наважку, прийнявши, що fE(Na2B4O7) =1/2; M(Na2B4O7) =381,4 г/моль. Готують розчин, як описано в 3.1. Особливістю виготовлення є те, що сіль треба розчиняти у гарячій воді. Після цього колбу охолоджують і лише тоді доводять об’єм розчину у мірній колбі до мітки. За формулою (3.12) обчислюють концентрацію одержаного розчину, C(Na2B4O7) або СЕ(Na2B4O7). Приготування розчину хлоридної кислоти. Розчин хлоридної кислоти готують приблизної концентрації з її концентрованого розчину. Для цього аре-ометром вимірюють густину розчину кислоти і за таблицею в довіднику визначають її приблизну молярну або масову концентрацію. У першому випад-ку за формулою (3.3), а в другому - за формулою (3.7) обчислюють об’єм концентрованої кислоти, потрібної для виготовлення 300 мл ~0,1 М розчину. Мірним циліндром відміряють обчислений об’єм концентрованої кислоти, поміщають його у пляшку об’ємом ~300 мл та доливають воду до верху. Закривають і старанно перемішують розчин до однорідності. Увага! Розчин концентрованої хлоридної кислоти необхідно розводити у витяжній шафі. Стандартизація розчину хлоридної кислоти. Взаємодія між розчинами хлоридної кислоти та натрію тетраборату відбувається за рівнянням, наведеним у 3.3.1. Виходячи із того, що на титрування використовують 0,05 М розчин тетраборату, та враховуючи розведення розчину в процесі титрування, можна вважати, що у точці еквівалентності є 0,1 М борна кислота, для якої pH обчислюють за формулою pH = Найбільш відповідний індикатор для титрування (табл. 3.1) - метиловий червоний. Якщо титрувати з метиловим оранжевим, то необхідною є сліпа про-ба, бо в цьому випадку витрачається деякий надлишок титранту. Сліпа проба - це виконання аналізу цим самим методом без визначуваного компонента з метою виявлення впливу фону на аналіз. Методика титрування. У дві конічні колби об’ємом 100-150 мл вносять піпеткою 10 мл розчину Na2B4O7, додають 1-2 краплі індикатора і титрують, перемішуючи, хлоридною кислотою до зміни забарвлення індикатора мети-лового червоного від жовтого до червоно-оранжевого. Виконують три-п’ять паралельних титрувань і за формулою (3.11) обчислюють концентрацію кисло-ти, C(HCl). Результати титрування (величини концентрацій розчину) оброб-ляють методами математичної статистики. Визначення натрію гідроксиду. При титруванні лугу хлоридною кисло-тою за реакцією H3O+ + OH- стрибок на кривій титрування є в межах pH 4-10, тому для титрування підхо-дить ряд індикаторів (табл. 3.1). Однак розчин лугу часто має домішки натрію карбонату, тому його ліпше титрувати з метиловим червоним. Методика титрування. У дві колби для титрування відбирають піпеткою 10 мл розчину лугу. Додають 1-2 краплі метилового червоного. Розчин лугу титрують розчином кислоти до зміни забарвлення від жовтого до червоно-оранжевого. Виконують три-п’ять титрувань та за об’ємом розчину HCl (формула (3.11)) і концентрацією C(HCl) обчислюють концентрацію лугу, C(NaOH). Результати аналізу обробляють методами математичної статистики. Визначення натрію карбонату. Титрування натрію карбонату хлорид-ною кислотою характеризується двома точками еквівалентності. При титруванні до першої точки еквівалентності перебігає реакція CO32- + H3O+ Як індикатор використовують фенолфталеїн, (табл. 3.1), оскільки pH розчину в першій точці еквівалентності має таке значення:
При титруванні до другої точки еквівалентності CO32+ + 2 H3O+ pH розчину має таке значення: pH = Методика титрування. Наважку солі масою ~1 г переносять у мірну колбу на 100 мл, розчиняють дистильованою водою, з якої попередньо видалено кип’ятінням CO2, та доводять водою до мітки (див. розд. 3.1). Піпеткою відбирають по 10 мл розчину і переносять у дві однакові колби. Титрують з метиловим оранжевим стандартизованим розчином HCl. За формулою (3.14) обчислюють масу натрію карбонату, m(Na2CO3) (M(Na2CO3)=105,99 г/моль, fE(Na2CO3) = 1/2)). Визначення кислот Кислоти визначають титруванням стандартизованим розчином лугу, який готують приблизної концентрації та стандартизують за первинним стандартом —розчином оксалатної кислоти (H2C2O4). Цим методом можна визначати сильні та слабкі кислоти, однак сіль амонію внаслідок малого значення константи протолізу ( Реагенти: H2C2O4 2H2O, перекристалізована; NaOH, концентрований розчин; HCl, ~0,1 M розчин; H3PO4, ~0,1 M розчин; індикатори: фенолфталеїн, метиловий червоний. Приготування розчину оксалатної кислоти. Оксалатна кислота (H2C2O4. 2H2O) у процесі зберігання втрачає частину кристалізаційної води, тому перед використанням її бажано перекристалізовувати. Слід також мати на увазі, що розчин оксалатної кислоти з часом може змінювати концентрацію. Готують первинний стандартний розчин кислоти об’ємом 200 або 250 мл з кон-центрацією 0,05 моль/л, як це описано в розд. 3.1. Для цього за формулою (3.12) обчислюють наважку кислоти (M(H2C2O4.2H2O)=126,07 г/моль, fE(H2C2O4)=1/2.). За наважкою (формула 3.12) обчислюють концентрацію розчину кислоти, C(H2C2O4) або CЕ(H2C2O4). Стандартизація розчину натрію гідроксиду. З концентрованого розчи-ну NaOH готують ~300 мл ~0,1 М розчину, аналогічно до описаного в розд. 3.3.1 приготування розчину хлоридної кислоти. Оксалатна кислота є протолітом середньої сили ( У точці еквівалентності розчин має лужну реакцію: pH = 7 + тому найкращим індикатором для титрування є фенолфталеїн (табл. 3.1). Методика титрування. У дві конічні колби на 100-150 мл переносять піпеткою 10 мл розчину H2C2O4, додають 1-2 краплі фенолфталеїну і титрують розчином лугу до появи блідо-рожевого забарвлення. Виконують три-п’ять титрувань і за об’ємом лугу обчислюють його концентрацію (формула (3.11)). Результати аналізу обробляють методами математичної статистики. Визначення хлоридної кислоти. При титруванні хлоридної кислоти 0,1 М розчином NaOH стрибок на кривій титрування значний (pH 4-10), тому можна використовувати різні індикатори: як фенолфталеїн, так і метиловий червоний (табл. 3.1). Методика титрування. У дві конічні колби на 100-150 мл переносять піпеткою 10 мл кислоти, додають 1-2 краплі індикатора і титрують до різкої зміни кольору (блідо-рожевого з фенолфталеїном, жовто-оранжевого з метило-вим червоним). Виконують три-п’ять титрувань. Знаючи об’єм розчину лугу, обчислюють концентрацію кислоти за формулою (3.11) (fE=1). Результати аналізу обробляють методами математичної статистики. Визначення фосфатної кислоти. Фосфатну кислоту, хоча вона три- протонна, можна титрувати лугом лише як одно- або двопротонну, бо константа протонізації за третім ступенем дуже мала (
другу точку еквівалентності (титрування до Na2HPO4, fE(H3PO4)=1/2) фіксують за фенолфталеїном або тимолфталеїном (див. табл. 3.1):
У цій роботі фосфатну кислоту визначають титруванням за першою та другою точкою еквівалентності. Методика титрування. У дві конічні колби для титрування піпеткою пере-носять по 10 мл фосфатної кислоти, додають 1-2 краплі метилового оран-жевого і титрують лугом до зміни рожевого забарвлення на жовте. Титрування проводять із врахуванням сліпої проби. Для цього відмірюють мірним цилін-дром 20 мл 0,33 М розчину NaH2PO4, додають 1 краплю метилового оранжевого і титрують лугом. Від об’єму лугу, який витратили на титрування H3PO4, віднімають об’єм лугу, витрачений на титрування сліпої проби. Виконують три-п’ять титрувань. За об’ємом розчину NaOH та його концентрацією обчис-люють концентрацію H3PO4 за формулою (3.11). Результати аналізу обробляють методами математичної статистики. У випадку титрування фосфатної кислоти до другої точки еквівалентності до такого ж об’єму кислоти, як зазначено вище, додають 5-7 крапель тимол-фталеїну і титрують лугом до блідо-голубого забарвлення. За об’ємом розчину NaOH (три-п’ять титрувань) та його концентрацією обчислюють концентрацію H3PO4 за формулою (3.11). Результати аналізу обробляють методами матема-тичної статистики. Визначення солей амонію. Прямимтитруваннямлугомсільамонію відтитрувати не можна, оскільки нема стрибка pH на кривій титрування, причи-ною чого є мале значення константи протолізу ( Метод зворотного титрування. У процесі нагрівання водних розчинів солей амонію з лугом виділяється аміак, NH4++ OH- який поглинають певним об’ємом титрованого розчину HCl. Залишок кислоти визначають титруванням розчином лугу. За іншим варіантом амонійну сіль обробляють надлишком титрованого розчину лугу та весь аміак видаляють кип’ятінням розчину. Надлишок NaOH, який не провзаємодіяв з іонами NH4+, відтитровують розчином HCl. За формулою (3.14) обчислюють масу NH4+ у розчині (для розчинів HCl і NaOH значення С і СЕ збігаються, бо fE =1) Метод заміщення (формальдегідний). У цьому методі у процесі взаємодії іонів амонію з формальдегідом виділяється еквівалентна кількість іонів H3O+, які відтитровують розчином лугу: 4 NH4+ + 6 HCOH = (CH2)6N4 + 2 H2O + 4 H3O+. Кількість лугу, яка витрачається на титрування, еквівалентна кількості амонію. Реагенти: NaOH, 0,1 М стандартизований розчин; HCl, 0,1 М стандартизований розчин; 20% розчин формальдегіду; індикатор фенолфталеїн. Методика титрування. У конічні колби для титрування переносять розчин солі амонію, додають 7 мл розчину формальдегіду і залишають на 1-2 хв, потім додають одну-дві краплі фенолфталеїну і титрують розчином натрію гідроксиду до блідо-рожевого забарвлення. Виконують два титрування. Обчислюють масу амонію m(NH4+) за формулою (3.14) (fE(NH4+) = fE(NaOH) = 1), підставляючи об’єм та концентрацію розчину лугу, який витрачається на титрування.
3.4. РЕДОКСИМЕТРІЯ (ОКИСНО - ВІДНОВНЕ ТИТРУВАННЯ) Окисно-відновні реакції використовують у титриметричному аналізі для визначення Феруму, Хрому, Мангану, Купруму, Ванадію та інших металів, а також деяких аніонів. Кількісною характеристикою перебігу цих реакцій є редокспотенціал E, який змінюється у процесі титрування. Характер цієї зміни має велике значення для встановлення точки еквівалентності, правильного вибору потрібного індика-тора, зміщення рівноваги реакції та ін. У точці еквівалентності для реакції a Red1+ b Ox2 Eт.е = де E Точку еквівалентності фіксують за допомогою окисно-відновних інди-каторів. Якщо робочий розчин інтенсивно забарвлений (наприклад KMnO4), то можливе безіндикаторне титрування. Механізм дії індикаторів різний. Це можуть бути речовини, які специфічно взаємодіють з однією із речовин, що беруть участь у реакції (SCN- з Fe2+; крохмаль з J2) або необоротно окисню-ються (метиловий оранжевий та метиловий червоний знебарвлюються внаслідок окиснення бромом). Окисно-відновні індикатори оборотно зміню-ють свій колір внаслідок окиснення або відновлення, взаємодіючи з титрантом, а окисна та відновна форми індикатора відрізняються забарвленням: IndOx + n e колір I колір II. Для таких індикаторів pT = E де E Підбирають індикатор так, щоб його pT збігався зі стрибком на кривій титрування; значення потенціалу в точці еквівалентності повинно бути близьке до E Таблиця 3.2.Характеристики деяких індикаторів методу редоксиметрії
Залежно від використовуваного титранту розрізняють декілька видів титрування: перманганатометричне, біхроматометричне, броматометричне, йодометричне та ін. 3.4.1. Перманганатометрія Метод перманганатометричного визначення грунтується на реакції окис-нення відновників калію перманганатом. Залежно від pH розчину MnO4- відновлюється до Mn (II), Mn (IV) або Mn(VI). У сильнокислому розчині процес відновлення описують таким рівнянням півреакції: MnO4- + 8 H3O+ + 5e У слабкокислому, нейтральному та слабколужному розчині іон MnO4- віднов-люється до Mn (IV): MnO4- + 4 H3O+ + 3e У сильнолужному середовищі MnO4- відновлюється до Mn (VI): MnO4- + e Оскільки півреакція відновлення іона MnO4- у сильнокислому середовищі характеризується найвищим значенням стандартного редокспотенціалу, то в методі перманганатометрії використовують саме цю реакцію, тобто проводять титрування у сильнокислому середовищі. Більшість визначень виконують у сульфатнокислому середовищі, якщо наявна хлоридна кислота, можливе част-кове окиснення хлорид-іонів перманганатом. У цьому випадку для зменшення дії спряженого окиснення іонів хлориду додають сіль Mn (II) або зв’язують іони Mn (III), які утворюються, у комплекс. Нітратна кислота, особливо та, що містить оксиди Нітрогену, непридатна, оскільки може спричинити деякі побічні процеси. Стандартизація розчину калію перманганату. Калій перманганат не відповідає вимогам, які ставлять до первинних стандартів. Препарат звичайно містить певну кількість домішок манган діоксиду. Крім того, концентрація розчину KMnO4 з часом зменшується внаслідок окиснення ним домішок орга-нічних речовин, що потрапляють у розчин як забруднення. Тому робочий розчин KMnO4 готують спочатку приблизної концентрації (CE~0,005 моль/л), а потім визначають його концентрацію за первинним стандартом (переважно H2C2O4.2 H2O, Na2C2O4, As2O3). Процес взаємодії іона перманганату з іоном оксалату складний: 2 MnO4- + 5 C2O42- + 16 H3O+ Він проходить декілька стадій з утворенням Mn (VI), Mn (IV), Mn (III) і їхніх оксалатних комплексів. Швидкість взаємодії невисока, але якщо є каталізатор Mn2+, реакція перебігає досить швидко з утворенням оксалатних комплексів Mn(III): MnC2O4+ (червоний), Mn(C2O4)2- (жовтий), Mn(C2O4)33- (червоний), між якими швидко встановлюється рівновага. Цей процес можна описати схемою MnO4- +4 Mn2++ 5n C2O42- +8 H3O+ Більшість авторів вважає, що ця реакція має декілька стадій. Спочатку утворюється манганат: MnO4- + MnC2O4 ® MnO42- + MnC2O4+. Утворений іон MnO42- у кислому розчині відновлюється іонами Mn2+ до Mn(III) за схемою Mn (VI) + Mn (II) ® 2 Mn (IV); Mn (IV) + Mn (II) ® 2 Mn (III). Ці комплекси повільно розкладаються з утворенням Mn2+ і CO2: Mn(C2O4)n(3-2n)+ На початку титрування, поки не утворився Mn2+, розчин KMnO4 знебар-влюється повільно. Після нагромадження в системі іонів Mn2+ знебарвлення прилитого розчину KMnO4 прискорюється. Тому для прискорення взаємодії іонів перманганату й оксалату розчин оксалату попередньо нагрівають до 70-80°С. У цій реакції fE(KMnO4) =1/5, fE(H2C2O4)= 1/2. Реагенти: H2C2O4. 2 H2O, перекристалізований; H2SO4 (1:4) розчин; KMnO4, ~ 0,01 М розчин. Методика визначення. Для визначення концентрації розчину KMnO4 використовують розчин H2C2O4 з концентрацією CE=0,05 моль/л, який попе-редньо виготовляють розчиненням у мірній колбі (див. розд. 3.1). Наважку обчислюють за формулою (3.12), M(H2C2O4. 2 H2O)=126,07 г/моль. Після приго-тування первинного стандартного розчину його концентрацію обчислюють за формулою (3.12) (fE(H2C2O4) = 1/2). Концентрацію KMnO4 визначають титруванням розчином H2C2O4. Піпеткою відбирають 10 мл розчину оксалатної кислоти та переносять його у конічну колбу на 100-150 мл (готують одночасно дві проби), доливають мірним циліндром 10 мл H2SO4 і нагрівають на піщаній бані до 70-80°С. Розчин не можна нагрівати до кипіння, бо тоді можливий розклад H2C2O4 ® CO2 + CO + H2O. До гарячого розчину H2C2O4 з бюретки додають декілька крапель розчину KMnO4 і, перемішуючи вміст колби, чекають, поки зникне рожеве забарвлення. Наступні порції калію перманганату додають швидше, але тільки після знебар-влення розчину. Титрування вважають закінченим, коли від останньої краплі прилитого розчину KMnO4 забарвлення розчину в колбі не зникає протягом 30с. Оскільки розчин KMnO4 забарвлений, то об’єм у бюретці вимірюють за верхнім краєм рідини. Концентрацію розчину KMnO4 обчислюють за формулою (3.11). Результати аналізу обробляють методами математичної статистики. Визначення Феруму. Суть методу полягає в окисненні Феруму (II) в кис-лому середовищі розчином KMnO4: MnO4- + 5 Fe2+ + 8 H3O+ Метод застосовують для визначення Феруму в рудах, шлаках, силікатах. Для переведення Феруму в розчин досліджувані матеріали обробляють кисло-тами або сплавляють з натрію карбонатом. Після такої обробки частина Феруму (II) може окиснитись до Феруму (III), тому перед титруванням Ферум (III) необ-хідно відновити. Як відновник переважно використовують станум (II) хлорид: 2 [FeCl4]- + [SnCl4]2- Надлишок стануму (II) хлориду окиснюють розчином HgCl2: [SnCl4]2- + 2 HgCl2 ® [SnCl6]2- + Hg2Cl2¯. Осад каломелі майже не окиснюється перманганатом. У випадку утворен-ня металічної ртуті (чорний осад), що може випасти при великому надлишку стануму (II), для визначення треба взяти нову порцію розчину Феруму (III) і повторити відновлення. Зіпсовану пробу з осадом металічної ртуті виливають в окрему посудину і добре її закривають корком. Ртуть не можна зливати у раковину! У процесі титрування концентрація іонів Феруму (III) збільшується і розчин поступово забарвлюється у жовтий колір, що заважає фіксації точки еквівалентності. Тому іони Феруму (III) зв’язують у безбарвний фосфатний комплекс, для чого перед титруванням до розчину додають фосфатну кислоту: Fe3+ + 2 H3PO4 + 3 H2O Реагенти: KMnO4, стандартизований розчин; H2SO4 (1:4) розчин; H3PO4, концентрований розчин; зразок, що містить Ферум (III). Методика визначення. За приблизним вмістом Феруму у зразку обчислю-ють його наважку з розрахунку на мірну колбу об’ємом 100 мл та концентра-цію 0,05 М (формула (3.13)). Розчиненням зразка готують розчин, як це описано в розд. 3.1, під час розчинення додають 20-25 мл H2SO4 (1:4) для запобігання гідролізу. Якщо після тривалого перемішування розчин є каламутним, то його обережно нагрівають на водяній бані. Після розчинення та охолодження розчин у мірній колбі доводять водою до мітки. Піпеткою відбирають аліквоту розчину (10 мл) та переносять у колбу для титрування, після чого додають 10-15 мл розчину H2SO4, 2-3 мл розчину H3PO4 і титрують розчином KMnO4 до появи рожевого забарвлення, яке не зникає протягом 20-30 с. Виконують три-п’ять титрувань. За даним кожного титру-вання за формулою (3.15) з урахуванням (3.18) обчислюють вміст Феруму у зразку (fE(KMnO4) = 1/5, fE(Fe) = 1). Результати обробляють методами матема-тичної статистики. Визначення Мангану (II). Калій перманганат окиснює іони Мангану (II) у нейтральному або слабколужному середовищі до MnO2: 2 MnO4- + 3 Mn2+ + 6 H2O Випадання бурого осаду (гідратованого манган діоксиду MnO(OH)2) ство-рює певні труднощі при визначенні кінця титрування, тому, виконуючи аналіз, потрібно дотримуватися рекомендованої методики. Як видно з рівняння реакції, зменшення концентрації іонів H3O+ у розчині, яке можна досягти введенням іонів OH-, тобто лугу, викликає зміщення рівно-ваги вправо. Однак надмірне збільшення концентрації іонів OH- може спричи-нити перебіг побічної реакції - окиснення Мангану (II) киснем повітря: Mn2+ + 2 OH- + 1/2 O2 Найсприятливішим середовищем для визначення Мангану (II) є розчин з pH 5-6. За цих умов майже не відбувається окиснення Мангану (II) киснем повітря, а концентрація іонів H3O+ є цілком достатньою для зміщення рівноваги реакції вправо. Ще однією особливістю перманганатометричного визначення Мангану є те, що MnO(OH)2 може утворювати солі з деякими основами або помітно адсорбувати катіони, зокрема і Мангану (II). Для видалення з осаду адсорбованих іонів у розчин додають іони Цинку (II), які витісняють іони Mn2+: Mn(MnO3) + Zn2+ ® Zn(MnO3) + Mn2+. Якщо у розчині є іони Fe3+, то рекомендують додавати цинк оксид, оскіль-ки останній осаджує іони Феруму: 2 Fe3+ + 3 ZnO + 3 H2O ® 2 Fe(OH)3¯ + 3 Zn2+. Титрування можна проводити, не відокремлюючи осаду феруму гідро-ксиду. Якщо немає солей Феруму, замість оксиду додають невелику кількість розчинної солі Цинку (ZnSO4, ZnCl2). Реагенти: KMnO4, стандартизований розчин, CE = 0,05 М; NH3, 2 М розчин; HCl, 2 М розчин; ZnO, водна суспензія; контрольний розчин солі Mn(II) Методика визначення. До одержаного кислого розчину, який містить 0,05-0,1 г Мангану, краплями додають водний розчин аміаку або натрію карбо-нату до появи каламуті, яка не зникає під час перемішування (ознака повної нейтралізації вільної кислоти). Одержану каламуть розчиняють у декількох краплях хлоридної кислоти, до прозорого розчину додають скаламучений з водою цинк оксид, доки на дні колби не почне утворюватися білий осад (над-лишок ZnO). Якщо є Ферум, виділяється також червоно-бурий осад феруму (III) гідроксиду. Підготовлений для титрування розчин розводять до 250-300 мл кип’ячою водою і, перемішуючи, по краплях додають до нього розчин калію перман-ганату. Кожну нову порцію KMnO4 додають лише після того, як осяде бурий осад MnO2 і розчин над осадом стане прозорим. Титрування вважають закінченим, коли розчин над осадом від додавання однієї краплі калію перман-ганату забарвиться у фіолетово-рожевий колір. Перше титрування дає лише орієнтовний результат. Під час другого і третього титрувань основну кількість калію перманганату доливають відразу, добре перемішуючи при цьому вміст колби. Решту розчину, приблизно 1 мл до закінчення титрування, додають по краплях. Під час титрування розчин весь час повинен бути нагрітим майже до кипіння. Обчислюючи результати, слід пам’ятати, що іон MnO4- відновлюється до MnO2, fE(KMnO4) = 1/3. Масу Мангану в розчині обчислюють за формулою (3.14), fE(Mn) = 1/2. Біхроматометрія В основі методу є реакція окиснення відновників іоном Cr2O72-, у резуль-таті якої відбувається відновлення Хрому (VI) до Хрому (III): Cr2O72- + 14 H3O+ + 6e ® 2 Cr3+ + 21 H2O. Окисно-відновний потенціал цієї півреакції значною мірою залежить від кислотності розчину:
(3.28) Отже, основною умовою біхроматометричного визначення є висока концен-трація іонів гідроксонію у розчині. Під час титрування розчин повинен бути 1 М відносно іонів гідроксонію. Солі Хрому (III), які утворюються, забарвлюють розчин у зелений колір з фіолетовим відтінком, тому, незважаючи на інтенсивне оранжеве забарвлення розчину K2Cr2O7, рекомендують застосовувати індикатор. Реагенти: K2Cr2O7, перекристалізований; H2SO4 (1:4) розчин; H3PO4, концентрований розчин; зразок, що містить Ферум; індикатор дифеніламін. Виготовлення стандартного розчину калію біхромату. Для очищення K2Cr2O7 від домішок його перекристалізовують, одержаний препарат вису-шують у сушильній шафі при 130°С і зберігають у закритій склянці. Висушена сіль не є гігроскопічною і її можна використовувати як первинний стандарт. Готують розчин K2Cr2O7 з концентрацією CE=0,05 моль/л, зважуючи сіль та розчиняючи її у мірній колбі на 200 або 250 мл (див. приготування первинного стандарту в розділі 3.1). Необхідне значення наважки та концентрацію одер-жаного розчину обчислюють за формулою (3.12), fE(K2Cr2O7) = 1/6. Визначення Феруму. В основі визначення Феруму (II) за калію біхрома-том є така реакція: Cr2O72- + 6 Fe2+ + 14 H3O+ Редокспотенціал у точці еквівалентності має значення:
Найкращий індикатор (табл. 3.2) - N-фенілантранілова кислота. Можна використовувати і дифеніламін. Обидва індикатори окиснюються н
|
|||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-26; просмотров: 583; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.169 (0.041 с.) |

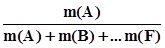 =
= 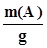 ; (3.1)
; (3.1)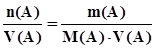 ; (3.2)
; (3.2)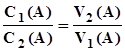 ; (3.3)
; (3.3) (або N(A)); (3.4)
(або N(A)); (3.4)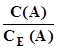 ; (3.5)
; (3.5)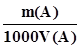 ; (3.6)
; (3.6)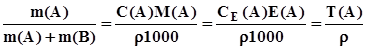 ; (3.7)
; (3.7)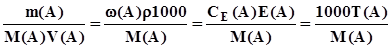 ; (3.8)
; (3.8)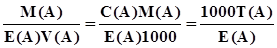 ; (3.9)
; (3.9) . (3.10)
. (3.10)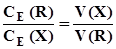 або
або 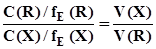 ; (3.11)
; (3.11)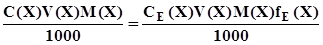 ; (3.12)
; (3.12)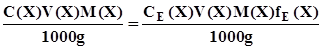 . (3.13)
. (3.13)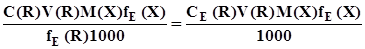 ; (3.14)
; (3.14)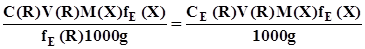 . (3.15)
. (3.15)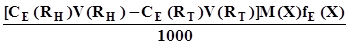 ; (3.16)
; (3.16) . (3.17)
. (3.17) . (3.18)
. (3.18)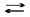 2 H2O.
2 H2O.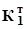 /
/ 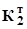 ³ 104 .
³ 104 .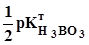 -
- 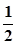 lg C(H3BO3) =
lg C(H3BO3) =  +
+ 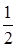 = 5,1. (3.20)
= 5,1. (3.20)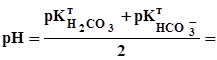
 8,35. (3.21)
8,35. (3.21)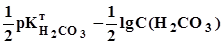 =
= 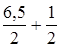 =3,9. (3.22) У цьому випадку для титрування використовують метиловий оранжевий.
=3,9. (3.22) У цьому випадку для титрування використовують метиловий оранжевий.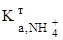 = 5,68.10-10) прямим титруванням не визначають.
= 5,68.10-10) прямим титруванням не визначають. = 6,5.10-2,
= 6,5.10-2,  = 6,1.10-5), її титрують розчином NaOH до середньої солі Na2C2O4.
= 6,1.10-5), її титрують розчином NaOH до середньої солі Na2C2O4. +
+ 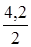 +
+  /
/ 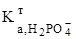 ³ 104;
³ 104;  =4,4. 10-13). Першу точку еквівалентності (титрування до NaH2PO4, fE(H3PO4)=1) фіксують за метиловим оранжевим:
=4,4. 10-13). Першу точку еквівалентності (титрування до NaH2PO4, fE(H3PO4)=1) фіксують за метиловим оранжевим: =
= 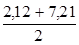 =4,7; (3.24)
=4,7; (3.24)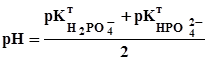 =
=  = 9,8. (3.25)
= 9,8. (3.25)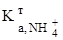 =5,68.10-10). Тому для визначення застосовують непрямі методи.
=5,68.10-10). Тому для визначення застосовують непрямі методи. , (3.26)
, (3.26) і E
і E 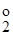 —стандартні потенціали першої та другої окисно-відновних півреакцій.
—стандартні потенціали першої та другої окисно-відновних півреакцій.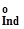 ±
± 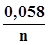 , (3.27),
, (3.27),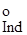 -стандартний електродний потенціал індикатора; n-кількість елек-тронів, які беруть участь у редокспроцесі.
-стандартний електродний потенціал індикатора; n-кількість елек-тронів, які беруть участь у редокспроцесі. = 1,51 В.
= 1,51 В. = 0,59 В.
= 0,59 В. = 0,56 В.
= 0,56 В. .
.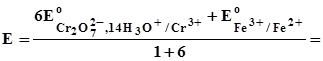
 . (3.29)
. (3.29)


