
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Гравіметричний та титриметричний аналізСодержание книги
Поиск на нашем сайте ГРАВІМЕТРИЧНИЙ ТА ТИТРИМЕТРИЧНИЙ АНАЛІЗ Для студентів хімічного факультету
Рекомендовано до друку кафедрою аналітичної хімії Протокол № 99 від 4.03.98
Львів ЛДУ 1998 Лабораторний практикум з аналітичної хімії. Гравіметричний та титриметричний аналіз/ Укл. Ю.Б.Кузьма, Я.Ф.Ломницька.- Львів: Ред.-вид. відділ Львів. ун-ту, 1998.- 55с.
У практикумі наведена коротка характеристика гравіметричного та титриметричного методів аналізу та деякі методики виконання, основні розрахункові формули. Для студентів хімічного факультету.
Редактор М.Ріпей
Cклали: Ю.Б. Кузьма, Я.Ф. Ломницька, П Е Р Е Д М О В А Особливість курсу аналітичної хімії, і кількісного аналізу зокрема, полягає в тому, що багато часу відведено лабораторному практикуму. Виконуючи лабо-раторні роботи, студенти повинні на практиці застосувати набуті теоретичні знання, освоїти певні хімічні операції, пов’язані з відокремленням та кількісним визначенням речовин. У практикумі стисло викладено роботи з гравіметричного і титриметрич-ного методів аналізу. Завдання підібрані так, щоб можна було урізноманітнити лабораторний практикум, пропонуючи студентам на вибір різні роботи. Значна увага приділена статистичній обробці результатів аналізу, наведені відповідні допоміжні таблиці, розглянуто приклади виконання розрахунків. Усе це сприя-тиме застосуванню методів математичної статистики для обчислення кінцевих результатів аналізу, особливо під час виконання робіт з титриметричного аналізу, в якому можна одержувати паралельні результати. Ці методичні вказівки можуть використовувати студенти хімічного, біологічного та геологічного факультетів університету. Виявлення сумнівного результату
Таблиця 1.1. Значення коефіцієнтів відбракування для n результатів та формули для обчислення сумнівних результатів за Q -тестом
Для виявлення сумнівного результату (промаху) обчислюють значення Q як частку від ділення різниці сумнівного результату і сусіднього з ним на різницю найбільшого та найменшого результатів (діапазон значень). Відповідні формули наведено в табл. 1.1. Якщо одержане число більше або дорівнює зна-ченню, наведеному у табл. 1.1, тобто Qексп ³ Q0,95, то цей результат відкидають. У випадку, коли одержали лише три результати, а один із них відкинули, треба виконати додаткове визначення, оскільки обчислювати середнє арифметичне за двома значеннями некоректно. Q-тест використовують, коли n ³ 3. Спочатку перевіряють найменше значення, а потім найбільше. Приклад. Виконали три паралельні визначення і з’ясували, що вміст ком-понента у зразку становить 30,13; 30,20; 31,23%. Необхідно визначити, який із результатів можна відкинути. Використовуючи Q-тест, найменший результат зберігають. Для найбільшого значення (31,23%) обчислюють частку Q: Qексп = Отримане значення Q дорівнює табличному Q0,95 (табл. 1.1), отже, сум-нівний результат треба відкинути та провести додатковий аналіз.
ГРАВІМЕТРИЧНИЙ АНАЛІЗ Гравіметричний метод аналізугрунтується на переведенні визначуваної речовини у важкорозчинну сполуку, яку відділяють, зважують та за її масою обчислюють вміст шуканого інгредієнта (елемента, речовини). Іноді замість осаду одержують летку речовину і після її повного видалення визначають змен-шення маси. Схема гравіметричного аналізу така: розчиняють наважку, діють 1,5-2,0-разовим надлишком осаджувача та одержують важкорозчинну сполуку (осаджувану форму). Осад відділяють фільтруванням, очищають промиванням (іноді перекристалізовують), висушують або прожарюють для одержання хіміч-но стійкої речовини сталого складу (гравіметрична форма). Точність аналізу залежить від раціонально вибраної осаджуваної форми (якомога менш розчинної), гравіметричної форми, умов осадження (концен-трації, pH розчину, температури, порядку змішування розчинів). Ці фактори впливають на повноту осадження та чистоту осаду. Важливим для проведення аналізу є правильно взята наважка. Відносна похибка гравіметричного аналізу не повинна перевищувати 0,1% (1.10-3 мас. частки), а похибка зважування на аналітичній вазі становить 1.10-4 г. Тоді приблизне значення маси гравіметричної форми обчислюють зі співвідношення
mосаду З огляду на масу гравіметричної форми для різних типів осадів запропо-новано різні маси осаджуваної форми (табл. 2.1).
Таблиця 2.1 Маса осаджуваної форми для різних типів осадів
Позначення та розрахункові формули, які використовують у гравіметричному методі аналізу: m(X) — м аса визначуваного інгредієнта (елемента, речовини), г; mграв — маса гравіметричної форми, г; g — маса наважки зразка, г; w(X) — масова частка інгредієнта у зразку; F(X) — фактор перерахунку маси осаду на масу інгредієнта; m(X) = mграв F(X); (2.2) w(X) = w(X) = Умови осадження аморфного та кристалічного осаду є різними. Для аморфних осадів осадження рекомендується проводити при високому перена-сиченні (швидке зливання гарячих концентрованих розчинів), після чого суспензію розводять водою для зменшення адсорбції. Кристалічні осади осад-жують з розведених розчинів і відстоюють під маточним розчином 4-24 год. Відділяють осад від розчину фільтруванням. Якщо осад кристалічний та осаджувана і гравіметрична форми збігаються, то його фільтрують через філь-трувальний тигель або фільтрувальну лійку з пористою скляною прокладкою, яка затримує осад, при пониженому тиску. В інших випадках для фільтрування використовують знезолені паперові фільтри (маса золи після спалювання менша від 0,0001 г). Дрібнозернисті кристалічні осади (BaSO4, CaC2O4) фільтрують через найщільніший фільтр (синя стрічка на пачці), більшість осадів - через фільтри середньої щільності (біла стрічка), аморфні - через менш щільні (червона, чорна стрічка). Для цього фільтр вміщують у скляну лійку. Проми-вають осад методом декантації, тобто чотири-п’ять разів у склянці порціями води (промивної рідини) по 20-30 мл, зливаючи кожну порцію на фільтр. Після завершення промивання весь осад переносять на фільтр. Після кількісного перенесення зі склянки завершують промивання осаду на фільтрі до негативної реакції на адсорбовані іони у двох-трьох краплях фільтрату. Способи переведення осаджуваної форми у гравіметричну такі: · висушування без нагрівання, для чого осад промивають спиртом, потім ефіром, залишок якого відсмоктують; · висушування з нагріванням до 110-120°С; · прожарювання при високих температурах (від 300-600 до 1000-1100°С), якщо необхідний термічний розклад вагової форми. Чисті тиглі і тиглі з осадом висушують (прожарюють) декілька разів, доки їхня маса не буде сталою. Для цього тиглі витримують у сушильній шафі чи муфельній печі при відповідній температурі 20-30 хв та охолоджують 15-20 хв у закритому ексикаторі, на дні якого є речовина, що адсорбує вологу. Посуд і матеріали для гравіметричного визначення: · порцеляновий тигель або скляний фільтруючий тигель (лійка); · склянка з термічно стійкого скла об’ємом 250-300 мл; · скляна паличка з гумовою насадкою на одному кінці; · скляна лійка; фільтрувальний папір; · ексикатор. Визначення Барію Осадження Барію проводять з розведеного нагрітого і підкисленого роз-чину, додаючи розчин сульфатної кислоти. Послідовність процесу осадження така, як осадження сульфату (див. розд. 2.1). Реагенти: H2SO4, 0,1 M розчин; HCl, 2 M розчин; AgNO3, 1% розчин. Методика визначення. До розчину, що містить Ba2+, додають воду до 100-110 мл, нагрівають до 60-70°С і підкислюють 2 мл розчину HCl. Осаджують Барій 2-разовим надлишком розчину H2SO4. Всі наступні операції виконують так, як це описано в розділі 2.1. Обчислюють масу m(Ba) за формулою (2.2), враховуючи що F(Ba)=
Визначення Феруму Ферум - один з найпоширеніших елементів у природі (руди, силікатні породи), він є в багатьох сплавах (чавуни, сталі та ін.). Із природних сполук найлегше розчинними є галуни, їх розчиняють у воді, підкисленій HNO3. Оксиди та сплави Феруму розчиняють у розчині хлоридної кислоти, після чого сполуки Феруму (II) окиснюють у сполуки Феруму (III) дією концентрованої HNO3. Якщо аналізують глини або силікати, то їх спочатку сплавляють з Na2CO3 та Na2S2O7, плав розчиняють у кислоті, одержаний розчин випарюють, при цьому випадає осад H2SiO3. Однак таким способом не вдається відділити Алюміній від Феруму, тому їх осаджують разом у вигляді гідроксидів. Визначенню Феруму у формі важкорозчинного Fe(OH)3 заважають Нікол (II), Кобальт (II), Цинк (II) та Манган (II) (останній окиснюється до MnO2), які теж утворюють осади гідрооксидів. З розчину солі Fe (II) дією водного розчину аміаку осаджують аморфний осад Fe(OH)3 ( Fe3+ + 3 NH3 + 3 H2O ® Fe(OH)3¯ + 3 NH4+ . Якщо для осадження використовують розчин лугу, то осад Fe(OH)3 адсор-бує помітну кількість лугу, тому метод придатний для відокремлення Fe3+, але не для його визначення. Якщо у розчині є іони Fe(II), то їх попередньо окиснюють дією Br2, H2O2, HNO3. Для одержання вагової форми Fe2O3 осад прожарюють у муфельній печі при 900°С: 2 Fe(OH)3 ® Fe2O3 + 3 H2O. Осад Fe2O3 не є гігроскопічним. Під час прожарювання необхідно забез-печити доступ повітря, оскільки при наявності відновників (CO, C з залишків фільтра), а також при дуже високій температурі (>1200°С) можливе часткове відновлення Феруму з утворенням Fe3O4. Реагенти: NH3, 10% розчин; HCl (1:1), або 20% розчин; HNO3, 70% розчин; AgNO3, 1% розчин. Методика визначення. Виходячи із приблизного вмісту Феруму у зразку та маси осаджуваної форми (табл. 2.1), обчислюють масу наважки за формулою (2.3). Зважують необхідну масу зразка. Для цього спочатку зважують пробірку з досліджуваним зразком на технічній, а потім на аналітичній вазі. Пересипають наважку у склянку, а пробірку з рештками речовини зважують на аналітичній вазі. За різницею обчислюють масу наважки. Зразок у склянці змочують 5-10 мл дистильованої води, додають 5-6 мл HCl і 3-4 мл HNO3 та нагрівають майже до кипіння. Про повноту окиснення свідчить стійке оранжеве забарвлення розчину (іноді темно-коричневе внаслідок утворення нестійкої сполуки FeCl2NO). Після завершення окиснення змивають внутрішні стінки склянки дистильованою водою. У склянку, помішуючи, дода-ють 5-10 мл розчину NH3 до виникнення відчутного запаху аміаку. Вміст склянки розводять до 100 мл гарячою дистильованою водою, перемішують і відстоюють 3-5 хв, після чого перевіряють повноту осадження, додаючи 2-3 краплі розчину NH3. Осад відфільтровують через фільтр середньої щільності (біла або рожева стрічка) та промивають методом декантації (три-чотири рази по 20 мл гарячою водою), після чого кількісно переносять на фільтр. Осад ще раз промивають на фільтрі. Відбирають 1-2 краплі фільтрату, в якому перевіряють наявність іонів хлориду. Фільтр з осадом на лійці висушують у сушильній шафі. Виймають фільтр, обережно, щоб не розсипати осад, складають, вміщують у порцеляновий тигель. Фільтр обзолюють на полум’ї пальника, після чого прожарюють при 900°С до одержання Fe2O3 (про що свідчить постійна маса зразка). За формулою (2.3) обчислюють вміст Феруму у зразку P(Fe) враховуючи, що: F(Fe) = Визначення Ніколу Нікол визначають у солях, мінералах та сплавах. Мінерали розкладають дією HNO3, залишок сплавляють з Na2CO3, після розчинення плаву у кислоті та наступного випарювання з кислотою осаджують H2SiO3. Сплави розчиняють у HCl, іони Феруму (II) окиснюють дією HNO3 у Ферум (III). Якщо є іони Феруму (III), Алюмінію (III), Титану (IV), які заважають визначенню, то їх зв’язують у стійкі розчинні комплекси тартратною або цитратною кислотою. Нікол (II) з розчину осаджують в аміачному середовищі спиртовим розчином диметилгліоксиму у формі легкого кристалічного осаду:
O-H...O-
Осад диметилгліоксимату Ніколу (Ni(HDm)2) дуже мало розчинний у воді, осаджується чистим, легко відфільтровується. Заважають визначенню деякі катіони VIII групи періодичної системи: Fe2+ утворює забарвлену розчинну сполуку; іони Паладію і Платини - нерозчинні сполуки; Co2+ попередньо осад-жують у формі K3[Co(NO2)6]. Осад диметилгліоксимату Ніколу фільтрують через фільтрувальний тигель (№ 3,4). Оскільки осаджувана і гравіметрична форма збігаються, то останню одержують шляхом висушування у сушильній шафі при 110-120°С. Можливе одержання гравіметричної форми NiO. У цьому випадку осад фільтрують через паперовий фільтр. Після його обзолення диметилгліоксимат Ніколу прожарюють при доброму доступі повітря та високій температурі. Недоліком є часткова сублімація ніколу диметилгліоксимату при 250°С. Реагенти: HCl (1:1) або 20% розчин; 2 М розчин; HNO3, концентрована; тартратна або цитратна кислота; NH3, 10% розчин; диметилгліоксим, 1% спиртовий розчин. Методика визначення. Осад ніколу диметилгліоксимату займає великий об’єм, тому треба вважати масу його такою ж, як і аморфного осаду (табл. 2.1). Знаючи вміст Ніколу у зразку, обчислюють наважку зразка (формула (2.3)). Визначення Ніколу у сталі. Розраховану наважку переносять у склянку та розчиняють у 10-15 мл розчину HCl, нагрівають, закривши склянку годин-никовим склом, доти, доки припиниться виділення водню. До розчину додають 3-5 мл концентрованої HNO3 для окиснення Феруму (II) та карбідів. Після закінчення виділення бурої пари NO2 скло знімають, споліскують дистильо-ваною водою, а розчин розводять до 100-120 мл водою. До розчину додають 5-7 г тартратної (цитратної) кислоти для зв’язування Fe3+. Після її розчинення додають розчин аміаку до появи запаху. Якщо з’явиться каламуть, то збіль-шують кількість тартратної кислоти. Після завершення цього етапу роботи додають у розчин розведену HCl до кислої реакції. Якщо з’явився невеликий осад, то його відфільтровують через нещільний фільтр та промивають гарячим розчином HCl, осаджують (див. осадження Ніколу). Визначення Ніколу у розчинних зразках. Наважку зразка переносять у склянку, розчиняють у 100 мл води при наявності 10 мл HCl та осаджують. Осадження Ніколу. До кислого розчину (містить 0,01-0,03 г Ni) додають 15-18 мл спиртового розчину диметилгліоксиму та нагрівають до 70-80°С (внаслідок тривалого нагрівання диметилгдіоксим починає розкладатися). До розчину краплями, помішуючи, додають розчин аміаку до появи слабкого запаху. Фільтрування починають через годину після випадання осаду, поперед-ньо перевіривши повноту осадження. Фільтрувальний тигель вставляють у шийку колби Бунзена, приєднують колбу до насоса. Фільтрують осад методом декантації, кількісно переносять його на фільтр та промивають гарячою водою до від’ємної реакції на іони Cl-. Осад просушують при 110-120°С. За масою ніколу диметилгліоксимату обчислюють вміст Ніколу у зразку w(Ni) за формулою (2.3), вважаючи, що F(Ni) =
3. ТИТРИМЕТРИЧНИЙ АНАЛІЗ Суть титриметричного методу аналізу полягає в тому, що розчин визна-чуваної речовини невідомої концентрації (X) взаємодіє із стехіометричною кількістю розчину реагенту (R): R + X За кількістю реагенту обчислюють кількість визначуваної речовини. У титриметричному аналізі використовують різні типи оборотних хімічних реакцій: кислотно-основні (протолітометрія); окиснення-відновлення (редокси-метрія); комплексоутворення; осадження. Процес поступового додавання роз-чину реагенту в розчин визначуваної речовини до досягнення стехіометричного співвідношення (точки еквівалентності) називається титруванням. У процесі титрування у точці еквівалентності кількість молів еквівалентів реагенту (R) дорівнює кількості молів еквівалентів визначуваної речовини (X). Точку стехіометричності (еквівалентності) фіксують хімічним методом за допомогою індикаторів (дуже зрідка без індикаторів) або фізико-хімічним методом (потенціометричне, кондуктометричне, амперометричне титрування). Механізм дії індикаторів є різним для різних типів реакцій. Визначення основ Концентрацію розчину основи визначають за допомогою титрування її робочим розчином кислоти. Для стандартизації робочого розчину кислоти як первинний стандарт використовують натрій тетраборат, Na2B4O7. 10 H2O, який взаємодіє з кислотою за рівнянням: B4O72- + 2 H3O+ + 3 H2O Реагенти: Na2B4O7. 10 H2O, перекристалізований; HCl, концентрований розчин; NaOH, ~0,1 М розчин; Na2CO3, ~0,1 М розчин; індикатори: метиловий оранжевий, метиловий червоний. Приготування розчину натрію тетраборату. Натрій тетраборат при зберіганні втрачає частину кристалізаційної води, тому перед приготуванням цю сіль потрібно перекристалізовувати. Для титрування використовують 0,05 М розчин, який готують у мірній колбі на 200 або 250 мл. За формулою (3.12) обчислюють необхідну наважку, прийнявши, що fE(Na2B4O7) =1/2; M(Na2B4O7) =381,4 г/моль. Готують розчин, як описано в 3.1. Особливістю виготовлення є те, що сіль треба розчиняти у гарячій воді. Після цього колбу охолоджують і лише тоді доводять об’єм розчину у мірній колбі до мітки. За формулою (3.12) обчислюють концентрацію одержаного розчину, C(Na2B4O7) або СЕ(Na2B4O7). Приготування розчину хлоридної кислоти. Розчин хлоридної кислоти готують приблизної концентрації з її концентрованого розчину. Для цього аре-ометром вимірюють густину розчину кислоти і за таблицею в довіднику визначають її приблизну молярну або масову концентрацію. У першому випад-ку за формулою (3.3), а в другому - за формулою (3.7) обчислюють об’єм концентрованої кислоти, потрібної для виготовлення 300 мл ~0,1 М розчину. Мірним циліндром відміряють обчислений об’єм концентрованої кислоти, поміщають його у пляшку об’ємом ~300 мл та доливають воду до верху. Закривають і старанно перемішують розчин до однорідності. Увага! Розчин концентрованої хлоридної кислоти необхідно розводити у витяжній шафі. Стандартизація розчину хлоридної кислоти. Взаємодія між розчинами хлоридної кислоти та натрію тетраборату відбувається за рівнянням, наведеним у 3.3.1. Виходячи із того, що на титрування використовують 0,05 М розчин тетраборату, та враховуючи розведення розчину в процесі титрування, можна вважати, що у точці еквівалентності є 0,1 М борна кислота, для якої pH обчислюють за формулою pH = Найбільш відповідний індикатор для титрування (табл. 3.1) - метиловий червоний. Якщо титрувати з метиловим оранжевим, то необхідною є сліпа про-ба, бо в цьому випадку витрачається деякий надлишок титранту. Сліпа проба - це виконання аналізу цим самим методом без визначуваного компонента з метою виявлення впливу фону на аналіз. Методика титрування. У дві конічні колби об’ємом 100-150 мл вносять піпеткою 10 мл розчину Na2B4O7, додають 1-2 краплі індикатора і титрують, перемішуючи, хлоридною кислотою до зміни забарвлення індикатора мети-лового червоного від жовтого до червоно-оранжевого. Виконують три-п’ять паралельних титрувань і за формулою (3.11) обчислюють концентрацію кисло-ти, C(HCl). Результати титрування (величини концентрацій розчину) оброб-ляють методами математичної статистики. Визначення натрію гідроксиду. При титруванні лугу хлоридною кисло-тою за реакцією H3O+ + OH- стрибок на кривій титрування є в межах pH 4-10, тому для титрування підхо-дить ряд індикаторів (табл. 3.1). Однак розчин лугу часто має домішки натрію карбонату, тому його ліпше титрувати з метиловим червоним. Методика титрування. У дві колби для титрування відбирають піпеткою 10 мл розчину лугу. Додають 1-2 краплі метилового червоного. Розчин лугу титрують розчином кислоти до зміни забарвлення від жовтого до червоно-оранжевого. Виконують три-п’ять титрувань та за об’ємом розчину HCl (формула (3.11)) і концентрацією C(HCl) обчислюють концентрацію лугу, C(NaOH). Результати аналізу обробляють методами математичної статистики. Визначення натрію карбонату. Титрування натрію карбонату хлорид-ною кислотою характеризується двома точками еквівалентності. При титруванні до першої точки еквівалентності перебігає реакція CO32- + H3O+ Як індикатор використовують фенолфталеїн, (табл. 3.1), оскільки pH розчину в першій точці еквівалентності має таке значення:
При титруванні до другої точки еквівалентності CO32+ + 2 H3O+ pH розчину має таке значення: pH = Методика титрування. Наважку солі масою ~1 г переносять у мірну колбу на 100 мл, розчиняють дистильованою водою, з якої попередньо видалено кип’ятінням CO2, та доводять водою до мітки (див. розд. 3.1). Піпеткою відбирають по 10 мл розчину і переносять у дві однакові колби. Титрують з метиловим оранжевим стандартизованим розчином HCl. За формулою (3.14) обчислюють масу натрію карбонату, m(Na2CO3) (M(Na2CO3)=105,99 г/моль, fE(Na2CO3) = 1/2)). Визначення кислот Кислоти визначають титруванням стандартизованим розчином лугу, який готують приблизної концентрації та стандартизують за первинним стандартом —розчином оксалатної кислоти (H2C2O4). Цим методом можна визначати сильні та слабкі кислоти, однак сіль амонію внаслідок малого значення константи протолізу ( Реагенти: H2C2O4 2H2O, перекристалізована; NaOH, концентрований розчин; HCl, ~0,1 M розчин; H3PO4, ~0,1 M розчин; індикатори: фенолфталеїн, метиловий червоний. Приготування розчину оксалатної кислоти. Оксалатна кислота (H2C2O4. 2H2O) у процесі зберігання втрачає частину кристалізаційної води, тому перед використанням її бажано перекристалізовувати. Слід також мати на увазі, що розчин оксалатної кислоти з часом може змінювати концентрацію. Готують первинний стандартний розчин кислоти об’ємом 200 або 250 мл з кон-центрацією 0,05 моль/л, як це описано в розд. 3.1. Для цього за формулою (3.12) обчислюють наважку кислоти (M(H2C2O4.2H2O)=126,07 г/моль, fE(H2C2O4)=1/2.). За наважкою (формула 3.12) обчислюють концентрацію розчину кислоти, C(H2C2O4) або CЕ(H2C2O4). Стандартизація розчину натрію гідроксиду. З концентрованого розчи-ну NaOH готують ~300 мл ~0,1 М розчину, аналогічно до описаного в розд. 3.3.1 приготування розчину хлоридної кислоти. Оксалатна кислота є протолітом середньої сили ( У точці еквівалентності розчин має лужну реакцію: pH = 7 + тому найкращим індикатором для титрування є фенолфталеїн (табл. 3.1). Методика титрування. У дві конічні колби на 100-150 мл переносять піпеткою 10 мл розчину H2C2O4, додають 1-2 краплі фенолфталеїну і титрують розчином лугу до появи блідо-рожевого забарвлення. Виконують три-п’ять титрувань і за об’ємом лугу обчислюють його концентрацію (формула (3.11)). Результати аналізу обробляють методами математичної статистики. Визначення хлоридної кислоти. При титруванні хлоридної кислоти 0,1 М розчином NaOH стрибок на кривій титрування значний (pH 4-10), тому можна використовувати різні індикатори: як фенолфталеїн, так і метиловий червоний (табл. 3.1). Методика титрування. У дві конічні колби на 100-150 мл переносять піпеткою 10 мл кислоти, додають 1-2 краплі індикатора і титрують до різкої зміни кольору (блідо-рожевого з фенолфталеїном, жовто-оранжевого з метило-вим червоним). Виконують три-п’ять титрувань. Знаючи об’єм розчину лугу, обчислюють концентрацію кислоти за формулою (3.11) (fE=1). Результати аналізу обробляють методами математичної статистики. Визначення фосфатної кислоти. Фосфатну кислоту, хоча вона три- протонна, можна титрувати лугом лише як одно- або двопротонну, бо константа протонізації за третім ступенем дуже мала (
другу точку еквівалентності (титрування до Na2HPO4, fE(H3PO4)=1/2) фіксують за фенолфталеїном або тимолфталеїном (див. табл. 3.1):
У цій роботі фосфатну кислоту визначають титруванням за першою та другою точкою еквівалентності. Методика титрування. У дві конічні колби для титрування піпеткою пере-носять по 10 мл фосфатної кислоти, додають 1-2 краплі метилового оран-жевого і титрують лугом до зміни рожевого забарвлення на жовте. Титрування проводять із врахуванням сліпої проби. Для цього відмірюють мірним цилін-дром 20 мл 0,33 М розчину NaH2PO4, додають 1 краплю метилового оранжевого і титрують лугом. Від об’єму лугу, який витратили на титрування H3PO4, віднімають об’єм лугу, витрачений на титрування сліпої проби. Виконують три-п’ять титрувань. За об’ємом розчину NaOH та його концентрацією обчис-люють концентрацію H3PO4 за формулою (3.11). Результати аналізу обробляють методами математичної статистики. У випадку титрування фосфатної кислоти до другої точки еквівалентності до такого ж об’єму кислоти, як зазначено вище, додають 5-7 крапель тимол-фталеїну і титрують лугом до блідо-голубого забарвлення. За об’ємом розчину NaOH (три-п’ять титрувань) та його концентрацією обчислюють концентрацію H3PO4 за формулою (3.11). Результати аналізу обробляють методами матема-тичної статистики. Визначення солей амонію. Прямимтитруваннямлугомсільамонію відтитрувати не можна, оскільки нема стрибка pH на кривій титрування, причи-ною чого є мале значення константи протолізу ( Метод зворотного титрування. У процесі нагрівання водних розчинів солей амонію з лугом виділяється аміак, NH4++ OH- який поглинають певним об’ємом титрованого розчину HCl. Залишок кислоти визначають титруванням розчином лугу. За іншим варіантом амонійну сіль обробляють надлишком титрованого розчину лугу та весь аміак видаляють кип’ятінням розчину. Надлишок NaOH, який не провзаємодіяв з іонами NH4+, відтитровують розчином HCl. За формулою (3.14) обчислюють масу NH4+ у розчині (для розчинів HCl і NaOH значення С і СЕ збігаються, бо fE =1) Метод заміщення (формальдегідний). У цьому методі у процесі взаємодії іонів амонію з формальдегідом виділяється еквівалентна кількість іонів H3O+, які відтитровують розчином лугу: 4 NH4+ + 6 HCOH = (CH2)6N4 + 2 H2O + 4 H3O+. Кількість лугу, яка витрачається на титрування, еквівалентна кількості амонію. Реагенти: NaOH, 0,1 М стандартизований розчин; HCl, 0,1 М стандартизований розчин; 20% розчин формальдегіду; індикатор фенолфталеїн. Методика титрування. У конічні колби для титрування переносять розчин солі амонію, додають 7 мл розчину формальдегіду і залишають на 1-2 хв, потім додають одну-дві краплі фенолфталеїну і титрують розчином натрію гідроксиду до блідо-рожевого забарвлення. Виконують два титрування. Обчислюють масу амонію m(NH4+) за формулою (3.14) (fE(NH4+) = fE(NaOH) = 1), підставляючи об’єм та концентрацію розчину лугу, який витрачається на титрування.
3.4. РЕДОКСИМЕТРІЯ (ОКИСНО - ВІДНОВНЕ ТИТРУВАННЯ) Окисно-відновні реакції використовують у титриметричному аналізі для визначення Феруму, Хрому, Мангану, Купруму, Ванадію та інших металів, а також деяких аніонів. Кількісною характеристикою перебігу цих реакцій є редокспотенціал E, який змінюється у процесі титрування. Характер цієї зміни має велике значення для встановлення точки еквівалентності, правильного вибору потрібного індика-тора, зміщення рівноваги реакції та ін. У точці еквівалентності для реакції a Red1+ b Ox2 Eт.е = де E Точку еквівалентності фіксують за допомогою окисно-відновних інди-каторів. Якщо робочий розчин інтенсивно забарвлений (наприклад KMnO4), то можливе безіндикаторне титрування. Механізм дії індикаторів різний. Це можуть бути речовини, які специфічно взаємодіють з однією із речовин, що беруть участь у реакції (SCN- з Fe2+; крохмаль з J2) або необоротно окисню-ються (метиловий оранжевий та метиловий червоний знебарвлюються внаслідок окиснення бромом). Окисно-відновні індикатори оборотно зміню-ють свій колір внаслідок окиснення або відновлення, взаємодіючи з титрантом, а окисна та відновна форми індикатора відрізняються забарвленням: IndOx + n e колір I колір II. Для таких індикаторів pT = E де E Підбирають індикатор так, щоб його pT збігався зі стрибком на кривій титрування; значення потенціалу в точці еквівалентності повинно бути близьке до E Таблиця 3.2.Характеристики деяких індикаторів методу редоксиметрії
Залежно від використовуваного титранту розрізняють декілька видів титрування: перманганатометричне, біхроматометричне, броматометричне, йодометричне та ін. 3.4.1. Перманганатометрія Метод перманганатометричного визначення грунтується на реакції окис-нення відновників калію перманганатом. Залежно від pH розчину MnO4- відновлюється до Mn (II), Mn (IV) або Mn(VI). У сильнокислому розчині процес відновлення описують таким рівнянням півреакції: MnO4- + 8 H3O+ + 5e У слабкокислому, нейтральному та
|
||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-26; просмотров: 484; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.137 (0.023 с.) |

 Часто, аналізуючи зразки, проводять лише три паралельні досліди. Щоб одержати оптимальний результат аналізу, використовують середнє арифме-тичне значення з трьох результатів. Однак трапляється, що два результати добре узгоджуються між собою, а третій відрізняється від двох попередніх. У таких випадках виникає бажання відкинути сумнівний результат і середнє значення обчислити за двома. Проте сумнівний результат для 3£ n £ 10 не треба відкидати, поки не буде обчислений Q-тест. Цей тест найчастіше обчислюють при 90% імовірності, а коефіцієнт відбракування позначають Q0,90. Якщо досто-вірна імовірність дорівнює 95%, то коефіцієнт відбракування буде Q0,95. У табл. 1.1. наведені значення коефіцієнтів відбракування для n результатів, розташованих у порядку зростання.
Часто, аналізуючи зразки, проводять лише три паралельні досліди. Щоб одержати оптимальний результат аналізу, використовують середнє арифме-тичне значення з трьох результатів. Однак трапляється, що два результати добре узгоджуються між собою, а третій відрізняється від двох попередніх. У таких випадках виникає бажання відкинути сумнівний результат і середнє значення обчислити за двома. Проте сумнівний результат для 3£ n £ 10 не треба відкидати, поки не буде обчислений Q-тест. Цей тест найчастіше обчислюють при 90% імовірності, а коефіцієнт відбракування позначають Q0,90. Якщо досто-вірна імовірність дорівнює 95%, то коефіцієнт відбракування буде Q0,95. У табл. 1.1. наведені значення коефіцієнтів відбракування для n результатів, розташованих у порядку зростання.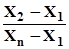 Qексп =
Qексп = 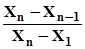

 =0,94.
=0,94. (2.1)
(2.1) г.
г. ; (2.3)
; (2.3) . (2.4)
. (2.4) =0,5884.
=0,5884. = 6,3.10-39):
= 6,3.10-39): = 0,6990.
= 0,6990.
 O-...H-O
O-...H-O


 CH3—C==N—OH CH3—C==N+ N==C—CH3
CH3—C==N—OH CH3—C==N+ N==C—CH3
 2 + Ni2++ 2 NH3® Ni +2 NH4+
2 + Ni2++ 2 NH3® Ni +2 NH4+
 CH3—C==N—OH CH3—C==N N+==C—CH3
CH3—C==N—OH CH3—C==N N+==C—CH3 = 0,2032.
= 0,2032. P.
P.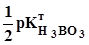 -
- 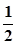 lg C(H3BO3) =
lg C(H3BO3) =  +
+ 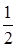 = 5,1. (3.20)
= 5,1. (3.20)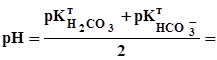
 8,35. (3.21)
8,35. (3.21)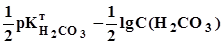 =
= 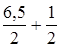 =3,9. (3.22) У цьому випадку для титрування використовують метиловий оранжевий.
=3,9. (3.22) У цьому випадку для титрування використовують метиловий оранжевий.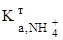 = 5,68.10-10) прямим титруванням не визначають.
= 5,68.10-10) прямим титруванням не визначають. = 6,5.10-2,
= 6,5.10-2,  = 6,1.10-5), її титрують розчином NaOH до середньої солі Na2C2O4.
= 6,1.10-5), її титрують розчином NaOH до середньої солі Na2C2O4. +
+ 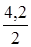 +
+  /
/ 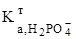 ³ 104;
³ 104;  =4,4. 10-13). Першу точку еквівалентності (титрування до NaH2PO4, fE(H3PO4)=1) фіксують за метиловим оранжевим:
=4,4. 10-13). Першу точку еквівалентності (титрування до NaH2PO4, fE(H3PO4)=1) фіксують за метиловим оранжевим: =
= 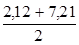 =4,7; (3.24)
=4,7; (3.24)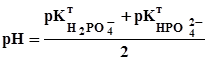 =
=  = 9,8. (3.25)
= 9,8. (3.25)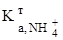 =5,68.10-10). Тому для визначення застосовують непрямі методи.
=5,68.10-10). Тому для визначення застосовують непрямі методи. , (3.26)
, (3.26)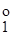 і E
і E 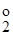 —стандартні потенціали першої та другої окисно-відновних півреакцій.
—стандартні потенціали першої та другої окисно-відновних півреакцій.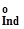 ±
± 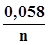 , (3.27),
, (3.27), -стандартний електродний потенціал індикатора; n-кількість елек-тронів, які беруть участь у редокспроцесі.
-стандартний електродний потенціал індикатора; n-кількість елек-тронів, які беруть участь у редокспроцесі. = 1,51 В.
= 1,51 В.


