
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Визначення Сульфуру у формі сульфатуСодержание книги
Поиск на нашем сайте
Сульфур може бути у формі сульфатів, розчинних у воді. Такий зразок розчиняють у воді з додаванням хлоридної кислоти. Якщо зразок містить спо-луки Феруму, то його попередньо відділяють, осаджуючи у формі Fe(OH)3. Якщо Сульфур є у формі сульфіду, то його попередньо окиснюють у SO42- сильним окисником при наявності кислоти: у мінералах - дією Br2+HNO3; сульфід у силікатах-сплавленням з Na2CO3+KNO3 при наявності кисню повітря; у сплавах та рудах-спалюванням у струмені кисню при високій температурі, після чого залишок обробляють пероксидом водню. Після переведення Сульфуру у розчинний сульфат його осаджують з гарячого розчину 1,5-разовим надлишком гарячого розчину BaCl2: Ba2+ + SO42- Одержують дрібнокристалічний осад, який витримують для агрегації 2-3 год, потім фільтрують через щільний фільтр (синя стрічка). Оскільки осад у воді досить розчинний ( Реагенти: BaCl2. 2 H2O, 5% розчин; HCl, 2 M розчин; AgNO3, 1% розчин. Методика визначення. До розчину, який містить SO42-, додають воду до об’єму 100-110 мл, нагрівають до 60-70°С і додають 2 мл розчину HCl. Для осадження додають краплями розчин BaCl2 (попередньо розчин BaCl2 розводять водою до 30-50 мл), безперервно помішуючи. Процес змішування повинен тривати не менше 10-15 хв. Осад з розчином відстоюють 2-3 год, перед фільтруванням перевіряють повноту осадження, додаючи ще дві-три краплі розчину BaCl2. Відфільтровують через щільний фільтр та промивають методом декантації. Після промивання осаду на фільтрі у фільтраті перевіряють, чи нема іонів Cl-. Після висушування фільтру з осадом фільтр виймають з лійки, складають і обзолюють у порцеляновому тиглі на полум’ї пальника. Після обзолення фільтру осад прожарюють при 800°С. Обчислюють масу сульфату m(SO4) за формулою (2.2), приймаючи, що
Визначення Барію Осадження Барію проводять з розведеного нагрітого і підкисленого роз-чину, додаючи розчин сульфатної кислоти. Послідовність процесу осадження така, як осадження сульфату (див. розд. 2.1). Реагенти: H2SO4, 0,1 M розчин; HCl, 2 M розчин; AgNO3, 1% розчин. Методика визначення. До розчину, що містить Ba2+, додають воду до 100-110 мл, нагрівають до 60-70°С і підкислюють 2 мл розчину HCl. Осаджують Барій 2-разовим надлишком розчину H2SO4. Всі наступні операції виконують так, як це описано в розділі 2.1. Обчислюють масу m(Ba) за формулою (2.2), враховуючи що F(Ba)=
Визначення Феруму Ферум - один з найпоширеніших елементів у природі (руди, силікатні породи), він є в багатьох сплавах (чавуни, сталі та ін.). Із природних сполук найлегше розчинними є галуни, їх розчиняють у воді, підкисленій HNO3. Оксиди та сплави Феруму розчиняють у розчині хлоридної кислоти, після чого сполуки Феруму (II) окиснюють у сполуки Феруму (III) дією концентрованої HNO3. Якщо аналізують глини або силікати, то їх спочатку сплавляють з Na2CO3 та Na2S2O7, плав розчиняють у кислоті, одержаний розчин випарюють, при цьому випадає осад H2SiO3. Однак таким способом не вдається відділити Алюміній від Феруму, тому їх осаджують разом у вигляді гідроксидів. Визначенню Феруму у формі важкорозчинного Fe(OH)3 заважають Нікол (II), Кобальт (II), Цинк (II) та Манган (II) (останній окиснюється до MnO2), які теж утворюють осади гідрооксидів. З розчину солі Fe (II) дією водного розчину аміаку осаджують аморфний осад Fe(OH)3 ( Fe3+ + 3 NH3 + 3 H2O ® Fe(OH)3¯ + 3 NH4+ . Якщо для осадження використовують розчин лугу, то осад Fe(OH)3 адсор-бує помітну кількість лугу, тому метод придатний для відокремлення Fe3+, але не для його визначення. Якщо у розчині є іони Fe(II), то їх попередньо окиснюють дією Br2, H2O2, HNO3. Для одержання вагової форми Fe2O3 осад прожарюють у муфельній печі при 900°С: 2 Fe(OH)3 ® Fe2O3 + 3 H2O. Осад Fe2O3 не є гігроскопічним. Під час прожарювання необхідно забез-печити доступ повітря, оскільки при наявності відновників (CO, C з залишків фільтра), а також при дуже високій температурі (>1200°С) можливе часткове відновлення Феруму з утворенням Fe3O4. Реагенти: NH3, 10% розчин; HCl (1:1), або 20% розчин; HNO3, 70% розчин; AgNO3, 1% розчин. Методика визначення. Виходячи із приблизного вмісту Феруму у зразку та маси осаджуваної форми (табл. 2.1), обчислюють масу наважки за формулою (2.3). Зважують необхідну масу зразка. Для цього спочатку зважують пробірку з досліджуваним зразком на технічній, а потім на аналітичній вазі. Пересипають наважку у склянку, а пробірку з рештками речовини зважують на аналітичній вазі. За різницею обчислюють масу наважки. Зразок у склянці змочують 5-10 мл дистильованої води, додають 5-6 мл HCl і 3-4 мл HNO3 та нагрівають майже до кипіння. Про повноту окиснення свідчить стійке оранжеве забарвлення розчину (іноді темно-коричневе внаслідок утворення нестійкої сполуки FeCl2NO). Після завершення окиснення змивають внутрішні стінки склянки дистильованою водою. У склянку, помішуючи, дода-ють 5-10 мл розчину NH3 до виникнення відчутного запаху аміаку. Вміст склянки розводять до 100 мл гарячою дистильованою водою, перемішують і відстоюють 3-5 хв, після чого перевіряють повноту осадження, додаючи 2-3 краплі розчину NH3. Осад відфільтровують через фільтр середньої щільності (біла або рожева стрічка) та промивають методом декантації (три-чотири рази по 20 мл гарячою водою), після чого кількісно переносять на фільтр. Осад ще раз промивають на фільтрі. Відбирають 1-2 краплі фільтрату, в якому перевіряють наявність іонів хлориду. Фільтр з осадом на лійці висушують у сушильній шафі. Виймають фільтр, обережно, щоб не розсипати осад, складають, вміщують у порцеляновий тигель. Фільтр обзолюють на полум’ї пальника, після чого прожарюють при 900°С до одержання Fe2O3 (про що свідчить постійна маса зразка). За формулою (2.3) обчислюють вміст Феруму у зразку P(Fe) враховуючи, що: F(Fe) = Визначення Ніколу Нікол визначають у солях, мінералах та сплавах. Мінерали розкладають дією HNO3, залишок сплавляють з Na2CO3, після розчинення плаву у кислоті та наступного випарювання з кислотою осаджують H2SiO3. Сплави розчиняють у HCl, іони Феруму (II) окиснюють дією HNO3 у Ферум (III). Якщо є іони Феруму (III), Алюмінію (III), Титану (IV), які заважають визначенню, то їх зв’язують у стійкі розчинні комплекси тартратною або цитратною кислотою. Нікол (II) з розчину осаджують в аміачному середовищі спиртовим розчином диметилгліоксиму у формі легкого кристалічного осаду:
O-H...O-
Осад диметилгліоксимату Ніколу (Ni(HDm)2) дуже мало розчинний у воді, осаджується чистим, легко відфільтровується. Заважають визначенню деякі катіони VIII групи періодичної системи: Fe2+ утворює забарвлену розчинну сполуку; іони Паладію і Платини - нерозчинні сполуки; Co2+ попередньо осад-жують у формі K3[Co(NO2)6]. Осад диметилгліоксимату Ніколу фільтрують через фільтрувальний тигель (№ 3,4). Оскільки осаджувана і гравіметрична форма збігаються, то останню одержують шляхом висушування у сушильній шафі при 110-120°С. Можливе одержання гравіметричної форми NiO. У цьому випадку осад фільтрують через паперовий фільтр. Після його обзолення диметилгліоксимат Ніколу прожарюють при доброму доступі повітря та високій температурі. Недоліком є часткова сублімація ніколу диметилгліоксимату при 250°С. Реагенти: HCl (1:1) або 20% розчин; 2 М розчин; HNO3, концентрована; тартратна або цитратна кислота; NH3, 10% розчин; диметилгліоксим, 1% спиртовий розчин. Методика визначення. Осад ніколу диметилгліоксимату займає великий об’єм, тому треба вважати масу його такою ж, як і аморфного осаду (табл. 2.1). Знаючи вміст Ніколу у зразку, обчислюють наважку зразка (формула (2.3)). Визначення Ніколу у сталі. Розраховану наважку переносять у склянку та розчиняють у 10-15 мл розчину HCl, нагрівають, закривши склянку годин-никовим склом, доти, доки припиниться виділення водню. До розчину додають 3-5 мл концентрованої HNO3 для окиснення Феруму (II) та карбідів. Після закінчення виділення бурої пари NO2 скло знімають, споліскують дистильо-ваною водою, а розчин розводять до 100-120 мл водою. До розчину додають 5-7 г тартратної (цитратної) кислоти для зв’язування Fe3+. Після її розчинення додають розчин аміаку до появи запаху. Якщо з’явиться каламуть, то збіль-шують кількість тартратної кислоти. Після завершення цього етапу роботи додають у розчин розведену HCl до кислої реакції. Якщо з’явився невеликий осад, то його відфільтровують через нещільний фільтр та промивають гарячим розчином HCl, осаджують (див. осадження Ніколу). Визначення Ніколу у розчинних зразках. Наважку зразка переносять у склянку, розчиняють у 100 мл води при наявності 10 мл HCl та осаджують. Осадження Ніколу. До кислого розчину (містить 0,01-0,03 г Ni) додають 15-18 мл спиртового розчину диметилгліоксиму та нагрівають до 70-80°С (внаслідок тривалого нагрівання диметилгдіоксим починає розкладатися). До розчину краплями, помішуючи, додають розчин аміаку до появи слабкого запаху. Фільтрування починають через годину після випадання осаду, поперед-ньо перевіривши повноту осадження. Фільтрувальний тигель вставляють у шийку колби Бунзена, приєднують колбу до насоса. Фільтрують осад методом декантації, кількісно переносять його на фільтр та промивають гарячою водою до від’ємної реакції на іони Cl-. Осад просушують при 110-120°С. За масою ніколу диметилгліоксимату обчислюють вміст Ніколу у зразку w(Ni) за формулою (2.3), вважаючи, що F(Ni) =
3. ТИТРИМЕТРИЧНИЙ АНАЛІЗ Суть титриметричного методу аналізу полягає в тому, що розчин визна-чуваної речовини невідомої концентрації (X) взаємодіє із стехіометричною кількістю розчину реагенту (R): R + X За кількістю реагенту обчислюють кількість визначуваної речовини. У титриметричному аналізі використовують різні типи оборотних хімічних реакцій: кислотно-основні (протолітометрія); окиснення-відновлення (редокси-метрія); комплексоутворення; осадження. Процес поступового додавання роз-чину реагенту в розчин визначуваної речовини до досягнення стехіометричного співвідношення (точки еквівалентності) називається титруванням. У процесі титрування у точці еквівалентності кількість молів еквівалентів реагенту (R) дорівнює кількості молів еквівалентів визначуваної речовини (X). Точку стехіометричності (еквівалентності) фіксують хімічним методом за допомогою індикаторів (дуже зрідка без індикаторів) або фізико-хімічним методом (потенціометричне, кондуктометричне, амперометричне титрування). Механізм дії індикаторів є різним для різних типів реакцій.
|
||||
|
|
Последнее изменение этой страницы: 2016-08-26; просмотров: 214; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.138.179.120 (0.009 с.) |

 BaSO4 ¯.
BaSO4 ¯. ), то його промивають невеликою кількістю води, а після останнього промивання перевіряють, чи є іони Ba2+. Якщо можливе співосадження іонів K+, Fe3+, то змінюють порядок зливання розчинів.
), то його промивають невеликою кількістю води, а після останнього промивання перевіряють, чи є іони Ba2+. Якщо можливе співосадження іонів K+, Fe3+, то змінюють порядок зливання розчинів.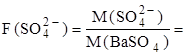 0,4150.
0,4150. =0,5884.
=0,5884. = 6,3.10-39):
= 6,3.10-39): = 0,6990.
= 0,6990.
 O-...H-O
O-...H-O

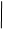

 CH3—C==N—OH CH3—C==N+ N==C—CH3
CH3—C==N—OH CH3—C==N+ N==C—CH3
 2 + Ni2++ 2 NH3® Ni +2 NH4+
2 + Ni2++ 2 NH3® Ni +2 NH4+
 CH3—C==N—OH CH3—C==N N+==C—CH3
CH3—C==N—OH CH3—C==N N+==C—CH3 = 0,2032.
= 0,2032.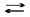 P.
P.


