
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Определение плотности твердых веществСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Для определения плотности твердых веществ применяют пикнометры, называемые волюмометрами. Емкость таких пикнометров обычно 50 мл. Вещество, относительную плотность которого нужно определить, должно не растворяться в растворителях. В качестве растворителей применяют этанол, хлороформ, керосин и другие растворители. Перед определением плотности твердого вещества его измельчают в фарфоровой ступке, высушивают в сушильном шкафу 1,5 — 2 ч при Относительную плотность твердого вещества начинают определять с относительной плотности выбранного растворителя. В волю-мометр, тщательно вымытый, высушенный и взвешенный на аналитических весах, помещают растворитель и снова взвешивают. В тот же волюмометр, также подготовленный к работе, насыпают несколько граммов исследуемого измельченного вещества, взвешивают, определяя навеску вещества. Затем наливают в волюмометр небольшими порциями растворитель. Содержимое волюмометра тщательно перемешивают встряхиванием. По заполнении волюмометра на 2/3 объема его помещают на 1 — 2 ч в водяную баню, нагретую до 60 — 65°С, для удаления пузырьков воздуха. Затем волюмометр охлаждают, доливают растворитель до метки и взвешивают, определяя массу волюмометра с измельченным веществом и растворителем. Относительную плотность твердого вещества определяют по уравнению:
где Правильно определить плотность твердого вещества можно только при условии полного удаления воздуха. 6.2. ОПРЕДЕЛЕНИЕ ВЯЗКОСТИ ЖИДКОСТИ В любой жидкости под влиянием внешней силы происходят взаимные перемещения молекул вещества. Возникающее при этом трение между молекулами, т.е. внутреннее сопротивление этому перемещению, называется внутренним трением, или вязкостью.
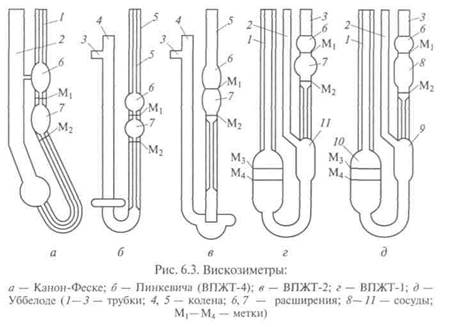
Наибольшее распространение при разных расчетах, а также при контроле качества получила кинематическая вязкость. Динамическую вязкость определяют в основном в научно-исследовательских работах. Вязкость существенно зависит от температуры, поэтому получаемое значение вязкости должно обязательно сопровождаться указанием температуры, при которой определялась вязкость. Приборы для определения вязкости называются вискозиметрами, схемы которых представлены на рис. 6.3. Чаще всего для определения кинематической вязкости пользуются стеклянными вискозиметрами типа Пинкевича (рис. 6.3, б) и В основе метода лежит известная формула Пуазейля динамической вязкости:
где
где р — плотность жидкости; g — ускорение силы тяжести; И — высота столба жидкости. Учитывая формулу
где величины вискозиметра (зависят только от его геометрических размеров). Обозначив
получим:
Величина С называется постоянной вискозиметра. Она не зависит от температуры и указывается в паспорте прибора. Для вискозиметров, находящихся постоянно в работе, постоянная С подвергается поверочной калибровке на истечение эталонной жидкости с известной кинематической вязкостью v3 и измерением времени ее истечения
Для определения вязкости необходимы: вискозиметр типа ВПЖТ-2 (см. рис. 6.3, в); термостатирующее устройство, обеспечивающее длительное поддержание заданной температуры с точностью ±0,03 °С при точных и ±0,1 "С при технических измерениях; термометр ртутный стеклянный с ценой наименьшего деления шкалы 0,05 °С для точных и 0,2 "С для технических измерений; секундомер; термостатирующая жидкость: дистиллированная вода, глицерин или смесь глицерина с водой в соотношении 1:1. Для определения кинематической вязкости вискозиметр подбирают таким образом, чтобы время истечения жидкости было не менее 200 с. Затем его тщательно промывают и высушивают. Пробу испытуемого вещества профильтровывают через бумажный фильтр. Вязкие жидкости перед фильтрованием нагревают до 50— 100 С. При наличии в жидкости воды ее осушают безводным сульфатом натрия или крупнокристаллической поваренной солью с последующим фильтрованием. В термостатирующем устройстве устанавливают требуемую температуру. Точность поддержания выбранной температуры имеет большое значение, поэтому термометр термостата должен быть установлен так, чтобы его резервуар оказался примерно на уровне середины капилляра вискозиметра с одновременным погружением всей шкалы. В противном случае вводится поправка на выступающий столбик ртути:
где (3 — коэффициент температурного расширения рабочей жидкости термометра (для ртутного термометра Р = 0,00016, для спиртового (3 = 0,001); h -т высота выступающего столбика рабочей жидкости термометра, выраженная в делениях шкалы термометра; t 1 — заданная температура в термостате, °С; t2 — температура окружающего воздуха вблизи середины выступающего столбика, °С. После этого наполняют вискозиметр ВПЖТ-2 испытуемой жидкостью, для чего на отводную трубку 3 надевают резиновую грушу. Затем, зажав пальцем колено 4, переворачивают вискозиметр и опускают колено 5 в сосуд с нефтепродуктом. С помощью груши всасывают пробу жидкости в вискозиметр до метки Mj. При этом необходимо следить, чтобы в капилляре и расширениях не было пузырьков воздуха, разрывов и пленок. В тот момент, когда уровень жидкости достигнет метки М,, вискозиметр вынимают из сосуда и быстро переворачивают в нормальное положение. Снимают резиновую грушу с отростка 3, обтирают колено 5 и надевают на него резиновую трубку. Наполненный вискозиметр устанавливают в термостатирующее устройство вертикально. До проведения отсчетов вискозиметр выдерживают в термостате 15 мин. Для проведения отсчетов грушей всасывают жидкость в колено 4 примерно до одной трети высоты расширения 7, после чего эту операцию прекращают. Жидкость под действием собственного веса начинает протекать через капилляр вниз. В тот момент, когда уровень жидкости достигнет метки М2 включают секундомер, а когда уровень достигнет метки М2, секундомер выключают. Время, отмеченное по секундомеру, записывают. Определение времени истечения повторяют несколько раз. Число измерений устанавливают в зависимости от времени истечения: пять измерений — при времени истечения от 200 до 300 с; четыре — от 300 до 600 с и три — при времени истечения свыше 600 с. При проведении отсчетов необходимо следить за постоянством температуры и отсутствием пузырьков воздуха. Для расчета вязкости определяют среднее арифметическое значение времени истечения. При этом учитывают только те отсчеты, которые отличаются не более чем на ±0,3 % при точных и на ±0,5 % при технических измерениях от среднего арифметического.
где С — постоянная вискозиметра, ческое учитываемых отсчетов времени истечения жидкости, с; g — ускорение свободного падения в месте измерения вязкости, м/с2; 9,807 — нормальное ускорение свободного падения, м/с2; К— коэффициент, учитывающий изменение гидростатического напора жидкости вследствие расширения ее при нагревании 6.3. ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ Способность вещества преломлять свет характеризуется показателем преломления. Если луч света пересекает границу раздела двух прозрачных сред, то направление луча изменяется в соответствии с законом преломления, согласно которому отношение синусов углов падения Каждая преломляющая свет среда характеризуется абсолютным показателем преломления п, который определяется как отношение скорости распространения света в вакууме к скорости его распространения в среде. В общем виде закон преломления света на границе двух сред может быть представлен соотношением:
где Постоянная для данной пары веществ величина п называется относительным показателем преломления и равна отношению их абсолютных показателей преломления пх и п2. Абсолютный показатель преломления вещества равен измеренному по отношению к воздуху, умноженному на абсолютный показатель преломления воздуха, который при нормальном атмосферном давлении и 20°С равен 1,00027. Показатель преломления зависит от длины волны падающего света и температуры среды. Например, обозначение С повышением температуры Если показатель преломления определяют прито по- лученную величину приводят к 20 °С по формуле:
где ломления при температуре опыта С показателем преломления связана величина, называемая удельной рефракцией
где Удельная рефракция зависит от внешних условий (температуры, давления) и агрегатного состояния вещества. В химическом анализе при решении ряда задач широко используют молекулярную рефракцию R, которая равна произведению удельной рефракции вещества
Молекулярная рефракция некоторых групп соединений зависит от природы и числа атомов в молекулах и может быть вычислена суммированием характерных для каждого атома, группы атомов или каждой химической связи констант — атомных рефракций или рефракций связей. Формула (6.34) связывает величины величины, четвертую можно найти вычислением по (6.34). Молекулярную рефракцию легко и достаточно точно вычисляют по хорошо известным атомным рефракциям и рефракциям связей. Поэтому уравнение (6.34) позволяет вычислить любую из величин — М или р, если две другие найдены экспериментально. Величину показателя преломления определяют на приборах — рефрактометрах, в которых фиксируют угол полного внутреннего отражения. Чаще всего для этого применяют универсальный рефрактометр марки УРЛ (рис. 6.4). Он дает возможность определять показатель преломления при длине волны желтой линии натрия 589,3 нм, пользуясь светом — либо дневным рассеянным, либо от матовой электрической лампы. С помощью рефрактометра УРЛ
можно определить показатель преломления в интервале от 1,3 до 1,7. Точность измерения 0,001. Основной рабочей частью рефрактометра (см. рис. 6.4) является камера 2, состоящая из двух, сложенных во время работы призм, соединенных шарниром, вокруг которого они могут вращаться. В закрытом состоянии обе призмы камеры удерживаются вместе специальным затвором. Каждая половинка камеры состоит из металлического кожуха, одну сторону которого образует плоскость вставленной в него стеклянной призмы. Между стеклянными призмами обеих половинок камеры, когда они сложены вместе, образуется очень тонкий зазор, в котором помещается исследуемое вещество. Камера может подключаться к термостату для поддержания постоянной температуры при измерении. Перед началом работы проверяют правильность установки шкалы показателей преломления измерительного блока. Для этого открывают затвор камеры, раскрывают обе половинки и протирают их мягкой тканью или ватой, смоченной эфиром. На поверхность нижней призмы наносят несколько капель свежепрокипяченной дистиллированной воды. Затем половинки складывают и закрывают затвор. Осветитель 3 поворачивают так, чтобы поле зрения было хорошо освещено и были видны на нем две пересекающиеся прямые (см. рис. 6.4, б). При этом нижняя часть поля зрения оказывается темной, а верхняя — светлой. Если граница окрашена, вращением маховика призмы дисперсного компенсатора б устраняют «окрашенность». Если на измерительной шкале граница светотени, проходя через точку пересечения прямых, соответствует 1,3330 — показателю преломления дистиллированной воды при 20°С, значит прибор настроен правильно. Если показатель преломления не соответствует этому числу, то рефрактометр юстируют согласно инструкции. После проверки показаний прибора приступают к измерению показателя преломления исследуемого вещества. Для этого предварительно поверхность призм протирают смоченной эфиром, а затем сухой ватой. Несколько капель исследуемого вещества наносят на поверхность измерительной призмы, при этом стараются не касаться поверхности призмы пипеткой. Осторожно опускают осветительную призму. В измерительную трубку наблюдают границу светотени и вращением маховика устраняют окрашенность. Точно совмещают границу светотени с точкой пересечения прямых и снимают данные по шкале. По окончании определения осветительную камеру поднимают и поверхность очищают. Измерения повторяют два-три раза. Если показатель преломления определяют при комнатной температуре, то полученную величину приводят к 20 "С, согласно формуле 6.32. 6.4. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ И КИПЕНИЯ Всякое твердое тело характеризуется свойственной ему кристаллической решеткой. В узлах решетки находятся образующие ее атомы или группы атомов, находящиеся в состоянии непрерывного колебательного движения, средняя кинетическая энергия которого зависит от температуры. При повышении температуры средняя энергия движения атомов или групп атомов увеличивается. Когда средняя энергия достигает определенной критической величины, кристаллическая решетка разрушается и твердое тело переходит в жидкое состояние, т. е. плавится. Температуру, при которой происходит этот процесс, называют температурой плавления. Каждое химически чистое индивидуальное вещество характеризуется присущей ему температурой плавления. Этот показатель является важной характеристикой чистоты вещества и его идентификации. Наиболее удобно (и обычно принято) определять температуру плавления в капилляре.
После охлаждения полученную капиллярную трубку нарезают острым напильником на отрезки 40—50 мм. С более узкого конца эти капилляры запаивают, для чего их вводят направленным вверх концом в пламя горелки и, вращая, непродолжительно нагревают. Для заполнения капилляра его вводят открытым концом в исследуемое вещество; при этом некоторое количество последнего падает в капилляр. Вещество перемещают на дно капилляра следующим образом. Тонкую стеклянную трубку (длиной 20 — 30 см) ставят в вертикальном положении на стол. В верхний конец трубки вносят капилляр и отпускают его. Падая, капилляр ударяется о крышку стола, в результате чего вещество спадает на дно и уплотняется там. Такой прием повторяют несколько раз. Для определения температуры плавления вещество должно заполнять капилляр слоем высотой 2—3 мм. Капилляр прикрепляют к термометру при помощи отрезка резиновой трубки шириной 1 мм. Столбик вещества должен находиться на уровне середины шарика термометра; резиновое кольцо должно охватывать верхний конец капилляра. Термометр с капилляром укрепляют в лапке штатива, как показано на рис. 6.5, и опускают в небольшой стакан. В стакан наливают вазелиновое масло и помещают магнитную мешалку. При необходимости нагрева до температуры выше 140 оС используют концентрированную серную кислоту. Так как горячая серная кислота может причинить тяжелые ожоги, то работать с ней надо осторожно Прибор нагревают на электрической плитке так, чтобы температура повышалась медленно. Если температура плавления вещества известна и ее определяют с целью установления степени чистоты вещества, то сначала быстро нагревают прибор до температуры, примерно на 10 °С ниже ожидаемой температуры плавления; затем скорость нагрева уменьшают и поднимают температуру очень медленно (не более чем на 1 °С в минуту). Температурой плавления считается та, при которой замечается первое появление жидкой фазы. Если вещество чистое, то оно полностью плавится при этой температуре ±(0,5... 1,0°С). Термометр, фиксирующий температуру плавления, должен быть предварительно проверен, так как периодическое нагревание и охлаждение термометра приводит к смещению нулевой точки, устанавливаемой при погружении его в смесь воды со льдом (полученном замораживанием дистиллированной воды). Точку, соответствующую 100 "С, проверяют, помещая термометр в пары кипящей воды. Если атмосферное давление при этом отличается от 760 мм рт. ст., то на каждые 10 мм рт. ст. вводят поправку в 0,37 °С. Точки вблизи 200 °С могут быть проверены помещением термометра в пары кипящего анилина (184,4 °С) или нафталина (218 °С). Поправка на колебание атмосферного давления в случае анилина равна 0,51 оС, в случае нафталина — 0,58 °С на каждые 10 мм рт. ст. Термометр дает правильные показания лишь в том случае, если весь столбик ртути нагрет до измеряемой температуры. Если ртутный столбик находится над поверхностью жидкости, он будет иметь более низкую температуру. Поэтому для установления истинной температуры плавления в показания термометра вводят поправку, рассчитываемую по формуле:
где At — температурная поправка, °С; к — дифференциальный коэффициент расширения ртути в стекле; п — длина, выступающая над жидкостью столбика ртути, выраженная числом градусов; Пример. Исследуемое вещество плавится при 230 °С. Длина выступающего столбика ртути 180 оС, его средняя температура 100 оС. Тогда поправка составляет 0,00016 • 180(230 - 100) = 3,7 "С или (округленно) 4°С. Следовательно, истинная температура плавления будет равна 230 + 4 = 234 оС. Она обозначается так: 234 °С (испр.).
Температуру плавления определяют не только с целью установления чистоты вещества, но и для его идентификации, т. е. установления тождества исследуемого соединения с каким-либо известным (описанным в литературе) веществом. Часто на основании представления о вероятном протекании реакции после предварительного ознакомления со свойствами и составом полученного вещества можно сделать предположение о его структуре. Тождество исследуемого вещества с описанным устанавливают на основании общности характерных реакций, совпадения состава и физических констант, из которых температура плавления имеет наибольшее значение, так как ее величина сильно изменяется даже при незначительных отличиях в строении веществ. Однако не всегда данные по определению температуры плавления могут считаться достаточными для идентификации двух веществ, так как не исключена возможность, что температуры плавления разных веществ могут оказаться одинаковыми или весьма близкими. В таких случаях прибегают к определению температуры плавления так называемой смешанной пробы обоих веществ. Для этого берут небольшое количество подлежащего идентификации вещества и сплавляют его с равным количеством чистого препарата того вещества, тождество с которым хотят установить. Полученный расплав измельчают, помещают в капилляр и обычным порядком определяют температуру его плавления. Если оба вещества идентичны, то их смесь будет плавиться при той же температуре, что и каждое из веществ в отдельности. В противном случае их смесь, как правило, плавится при более низкой температуре, чем чистые вещества. Температуру кипения обычно определяют при перегонке вещества в процессе его очистки. Для получения более точных данных исследуемое вещество перегоняют из перегонной колбы, используя проверенный термометр. Удобно пользоваться набором термометров с укороченной шкалой, так как при этом отпадает необходимость вводить поправку на выступающий над пробкой столбик ртути. В колбу обязательно нужно поместить капилляры или кусочки керамики для устранения перегрева жидкости и обеспечения равномерности кипения. Необходимо следить за тем, чтобы не нагревались не покрытые жидкостью стенки колбы, так как при этом возможен перегрев паров кипящей жидкости, и термометр будет показывать более высокую температуру. Если нагревание ведут на открытом пламени горелки, то колбу следует вставить в круглое отверстие, вырезанное в куске асбестового картона. Диаметр этого отверстия должен быть несколько меньше (около 3/4) диаметра колбы. Определение температуры кипения малого количества жидкости удобно проводить микрометодом Сиволобова. Каплю жидкости помещают в запаянную с одного конца тонкостенную стеклянную трубку диаметром 2,5 — 3,0 мм. В трубку опускают запаянный с верхнего конца капилляр, прикрепляют трубку к термометру (рис. 6.6) и нагревают в приборе для определения температуры плавления. Как только исследуемая жидкость в капилляре нагреется до температуры чуть выше температуры ее кипения (перегрев), из капилляра непрерывной струей В последнее время для точного определения температур плавления и кипения используют современные полуавтоматические приборы, как правило, снабженные встроенным увеличителем (регулируемой линзой), микропроцессорным контролем нагрева печи и считывания показаний, памятью для хранения результатов определения. Нагревающий блок может содержать несколько капилляров разного диаметра. Скорость нагрева образца задают в зависимости от требуемой точности определения. Приборы могут иметь цифровой дисплей с разрешением до 0,1 °С и разной точностью измерения. В качестве индикатора температуры используют либо откалиброванный термометр, либо термопару. Контрольные вопросы 1. Дайте определение абсолютной и относительной плотности. Приве 2. Расскажите об определении плотности жидких веществ с помощью 3. Приведите уравнения для пересчета плотности на стандартную тем 4. Дайте определение плотности твердых веществ. 5. Расскажите о динамической, кинематической, удельной и условной 6. Как измерить вязкость жидкостей с помощью вискозиметра, посто 7. Дайте определение показателя преломления, удельной и молекуляр 8. Дайте определение показателя преломления. 9. Как измеряют температуры плавления и кипения?
7.1. ОСНОВНЫЕ ВИДЫ ПРОБ Проба — наиболее репрезентативная часть исследуемого объекта. Отбор пробы является важной частью анализа, необходимым условием получения правильных результатов. Ошибки, возникающие из-за неправильного отбора проб, в дальнейшем исправить невозможно. Особую трудность вызывает отбор представительной пробы, состав которой соответствует составу анализируемого объекта. Реально отобранные для анализа пробы по составу, в большей или меньшей степени, отличаются от анализируемого объекта, что и является основной причиной погрешностей анализа. Условия, которые следует соблюдать при отборе проб, настолько разнообразны, что нельзя дать подробных рекомендаций для всех случаев в соответствии со всеми требованиями. В конкретных условиях следует руководствоваться целями исследования и общими принципами, регламентирующими отбор проб для анализов. Эти принципы регламентируются ГОСТами для каждой конкретной методики анализа. В соответствии с целями анализа проводят разовый или серийный отбор проб. При разовом отборе пробу берут один раз и рассматривают результаты одного анализа. Этот способ применяется редко, когда результатов единичного анализа достаточно для суждения о составе и качестве исследуемого объекта. Часто состав объекта изменяется в зависимости от места и времени отбора пробы, в этих случаях проводят серийный отбор проб. При анализе серии взятых проб определяется изменение содержания отдельных компонентов с учетом места, времени отбора или обоих этих факторов. Полученные результаты обрабатываются статистически. Типичным примером серийного отбора проб является зональный отбор. Другой распространенный метод серийного отбора проб — отбор через определенные промежутки времени, позволяющий следить за изменением состава объекта во времени. Различают пробы двух основных видов — простую и смешанную. Простую пробу получают однократным отбором всего требуемого количества объекта. Анализ простой пробы дает сведения о составе объекта в данный момент в данном месте. Смешанную пробу получают, смешивая простые пробы, взятые в одном и том же месте через определенные промежутки времени или отобранные одновременно в разных местах. Эта проба характеризует средний состав исследуемого объекта или средний состав за определенный период времени (за час, смену, день и т.д.), или, наконец, средний состав с учетом как места, так и времени. Смешанную пробу нельзя отбирать за период больше одних суток. При необходимости более длительного хранения пробу консервируют. Смешанную пробу нельзя использовать для определения тех компонентов и характеристик, которые изменяются во времени (растворенные газы, рН и т.д.). Эти определения проводят в каждой составляющей пробы отдельно. Количество пробы, которое необходимо отобрать, зависит от числа определяемых компонентов. 7.2. ОТБОР ПРОБ ГАЗООБРАЗНЫХ ВЕЩЕСТВ (ВОЗДУХА) Одной из наиболее актуальных задач газового анализа является определение содержания газообразных веществ в воздухе. В связи с тем, что анализ газообразной фазы зачастую более сложен как в аппаратурном, так и методическом оформлении, чем анализ жидких и твердых объектов, то крайне ответственным этапом является правильный отбор пробы. Правильность отбора пробы газообразных веществ определяется не только тщательностью технического выполнения операции, но учетом ряда важных факторов, например, физико-химических свойств улавливаемой примеси, соответствия скорости и отбираемого объема пробы составу поглотительного раствора и чувствительности применяемой реакции. Необходимо подчеркнуть, что в случае анализа воздуха отбор пробы имеет особое значение, так как он непосредственно связан с гигиенической оценкой воздушной среды и проведением сани-тарно-технических мероприятий. От того, будет ли, например, отбор пробы продолжительным или кратковременным, зависит суждение о степени опасности изучаемого производства, если учесть, что воздушная среда является подвижной системой, а поступление вредных веществ может происходить как прерывисто, так и монотонно. При длительном отборе пробы результат получается усредненным за данный отрезок времени. При кратковременном отборе пробы состав газовой фазы может резко меняться. Для исключения случайной погрешности требуется проводить динамический отбор проб. Для определения содержания компонентов газовой фазы известный объем последней пропускают с определенной скоростью через специальные поглотительные приборы. В качестве поглощающих веществ используют растворы соответствующих органических и неорганических соединений, связывающие определяемый
компонент при его прохождении через раствор. Наиболее широкое распространение в лабораторной практике газового анализа получили поглотители Зайцева, Полежаева, Яворской и поглотитель с фильтровальной пластиной (рис. 7.1). Поглотители Зайцева, Полежаева и Яворской разнятся поглотительной способностью и используются для определения разных веществ в газовой фазе. Оптимальная скорость аспирации через эти приборы составляет в среднем 0,5 л/мин. Быстрый и эффективный способ отбора проб — аспирация через «кипящий» слой сорбента, небольшое сопротивление которого позволяет доводить скорость аспирации до 20 л/мин. Для этой цели предложен поглотительный прибор, изображенный на рис. 7.1, г. В нижнюю часть колонки впаяна стеклянная пористая пластинка, поддерживающая слой сорбента в неподвижном состоянии. Точность химического анализа состава газовой фазы в значительной степени зависит от правильности измерения ее расхода при прохождении через поглотительный сосуд. Для этого используют приборы, называемые расходомерами. С их помощью можно с достаточной точностью определить объем газа, прошедшего через поглотитель. 7.3. ОТБОР ПРОБ ЖИДКОСТИ Способ отбора жидкой пробы зависит от цели, которая ставится перед исследователем в каждом отдельном случае. Необходимо всегда следить за тем, чтобы отбираемая проба не оказалась случайной. Если состав жидкой фазы меняется, то перед отбором пробы необходимо подробно изучить этот процесс и отбирать для анализа средние или среднепропорциональные пробы через определенное время. Средняя проба должна быть составлена из равных коли- честв жидкости, взятой через равные интервалы времени. И средние, и среднепропорциональные пробы обычно берут в течение суток, сливая отдельные порции в большие чисто вымытые бутыли. По истечении суток содержимое бутыли тщательно перемешивают и для анализа отливают часть жидкости в чисто вымытую посуду. Для характеристики изменения состава жидкости со временем необходимо отбирать разовые пробы и определять в них отдельные компоненты, характерные для данного стока. Такие пробы следует брать, например, через каждые 1 —2 ч, а иногда и через несколько минут (в зависимости от цели исследования) и сразу анализировать отобранные пробы. В случае отбора проб воды из технологических стоков отбирать пробу лучше всего при помощи батометра или склянки емкостью 1,0—1,5 л, заключенной в металлическую оправу со свинцовым дном. Склянку закрепляют в металлической оправе специальным зажимом, охватывающим ее горлышко, и закрывают резиновой пробкой, привязанной к шнуру, при помощи которого пробку можно вынуть на требуемой глубине. Если в воде присутствует свободный сероводород, на правильный отбор пробы должно быть обращено особое внимание. В таких случаях следует на месте отбирать специальные пробы воды только для определения сероводорода. Каждую пробу отби
|
|||
|
|
Последнее изменение этой страницы: 2016-04-19; просмотров: 2357; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.156 (0.017 с.) |



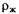 — относительная плотность жидкости;
— относительная плотность жидкости;  — масса измельченного вещества, навески, г;
— масса измельченного вещества, навески, г;  — масса пикнометра, наполненного растворителем, г;
— масса пикнометра, наполненного растворителем, г;  — масса пикнометра с измельченным веществом и растворителем, г.
— масса пикнометра с измельченным веществом и растворителем, г.
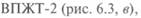 с помощью которых измеряют кинематическую вязкость прозрачных жидкостей при положительных и отрицательных значениях температуры.
с помощью которых измеряют кинематическую вязкость прозрачных жидкостей при положительных и отрицательных значениях температуры.
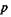 — давление, при котором происходит истечение жидкости из капилляра;
— давление, при котором происходит истечение жидкости из капилляра; 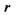 — радиус капилляра;
— радиус капилляра; 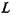 — длина капилляра;
— длина капилляра; 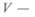 объем жидкости, протекающей через капилляр;
объем жидкости, протекающей через капилляр; 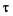 — время истечения жидкости в объеме
— время истечения жидкости в объеме 

 При определении кинематической вязкости жидкость протекает через капилляр под действием собственного веса, его можно выразить формулой:
При определении кинематической вязкости жидкость протекает через капилляр под действием собственного веса, его можно выразить формулой:
 получим:
получим:
 имеют постоянное значение для данного
имеют постоянное значение для данного






 Кинематическую вязкость испытуемой жидкости при температуре t вычисляют по формуле:
Кинематическую вязкость испытуемой жидкости при температуре t вычисляют по формуле:
 среднее арифмети-
среднее арифмети- где
где  — разность между температурой определяемой жидкости при заполнении вискозиметра и его температурой при определении вязкости).
— разность между температурой определяемой жидкости при заполнении вискозиметра и его температурой при определении вязкости). и преломления
и преломления 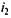 есть постоянная величина и обозначается символом п.
есть постоянная величина и обозначается символом п.
 — угол падения луча;
— угол падения луча; 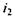 — угол преломления луча при переходе из первой среды во вторую.
— угол преломления луча при переходе из первой среды во вторую.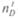 означает, что показатель преломления соответствует длине волны желтой линии натрия
означает, что показатель преломления соответствует длине волны желтой линии натрия  и температуре
и температуре  Определяют показатель преломления чаще всего, используя эту длину волны проходящего света, при 20°С, обозначая показатель преломления
Определяют показатель преломления чаще всего, используя эту длину волны проходящего света, при 20°С, обозначая показатель преломления 
 уменьшается, а с понижением — увеличивается. Среднее изменение показателя преломления при изменении температуры на 1 °С называется температурным коэффициентом
уменьшается, а с понижением — увеличивается. Среднее изменение показателя преломления при изменении температуры на 1 °С называется температурным коэффициентом 


 — показатель преломления при
— показатель преломления при  — показатель пре-
— показатель пре- — температурный коэффициент.
— температурный коэффициент. вычисляемая по уравнению Лоренца:
вычисляемая по уравнению Лоренца:
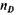 — относительный показатель преломления, найденный экспериментально;
— относительный показатель преломления, найденный экспериментально; 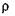 — плотность вещества, определенная при той же температуре, что и
— плотность вещества, определенная при той же температуре, что и 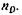
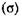 на его молярную массу (М):
на его молярную массу (М):
 Зная три
Зная три





 Для приготовления капилляров берут широкую тонкостенную стеклянную трубку, тщательно моют ее дистиллированной водой и высушивают. Высушенную трубку нагревают (непрерывно вращая ее) на пламени паяльной горелки до размягчения, затем быстро вынимают из огня и вытягивают до получения капилляра диаметром около 1 мм.
Для приготовления капилляров берут широкую тонкостенную стеклянную трубку, тщательно моют ее дистиллированной водой и высушивают. Высушенную трубку нагревают (непрерывно вращая ее) на пламени паяльной горелки до размягчения, затем быстро вынимают из огня и вытягивают до получения капилляра диаметром около 1 мм. (в предохранительных очках).
(в предохранительных очках).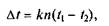
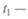 температура жидкости; t2 — средняя температура выступающего столбика (определяется при помощи вспомогательного термометра, шарик которого прикладывают к середине выступающего ртутного
температура жидкости; t2 — средняя температура выступающего столбика (определяется при помощи вспомогательного термометра, шарик которого прикладывают к середине выступающего ртутного 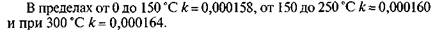 столбика).
столбика).

 Серная кислота в приборе для определения температуры плавления со временем темнеет вследствие попадания органических загрязнений. Это потемнение легко устранить прибавлением небольшого кристаллика селитры. Во избежание загрязнения серной кислоты нужно следить, чтобы она не смачивала резиновое колечко, прикрепляющее капилляр к термометру.
Серная кислота в приборе для определения температуры плавления со временем темнеет вследствие попадания органических загрязнений. Это потемнение легко устранить прибавлением небольшого кристаллика селитры. Во избежание загрязнения серной кислоты нужно следить, чтобы она не смачивала резиновое колечко, прикрепляющее капилляр к термометру. начнут выходить пузырьки. Для точного установления температуры кипения дальнейшее нагревание прекращают и отмечают ту температуру, при которой перестанут выделяться пузырьки.
начнут выходить пузырьки. Для точного установления температуры кипения дальнейшее нагревание прекращают и отмечают ту температуру, при которой перестанут выделяться пузырьки.
 Глава 7 ПРОБООТБОР
Глава 7 ПРОБООТБОР





