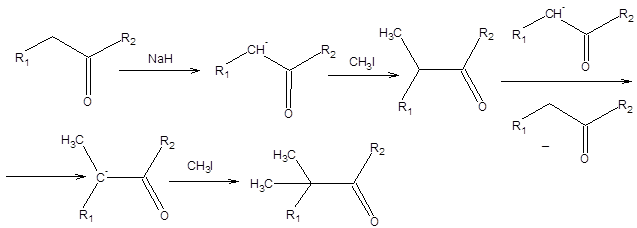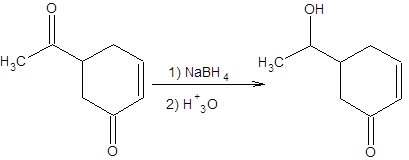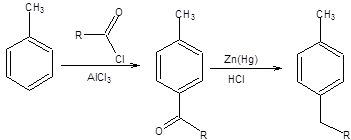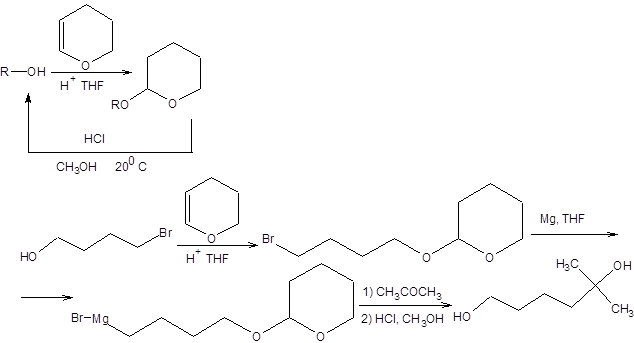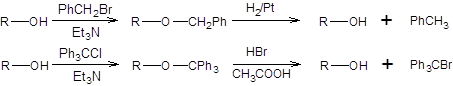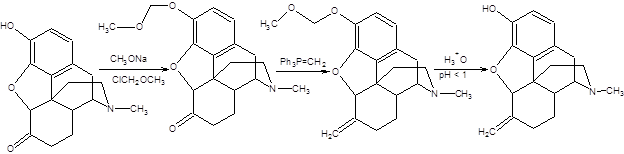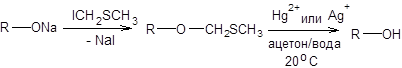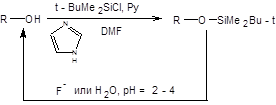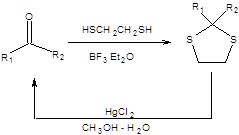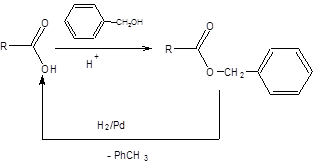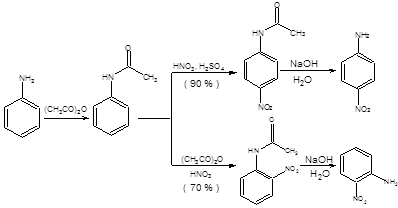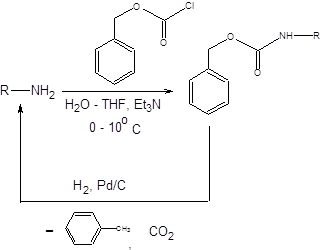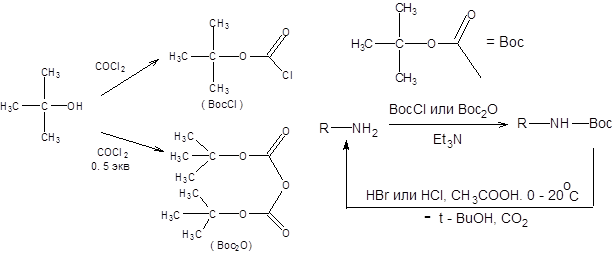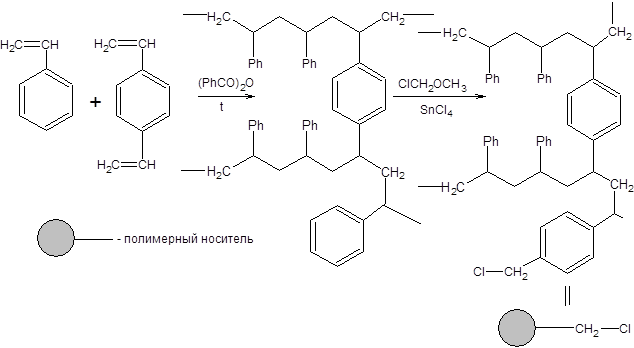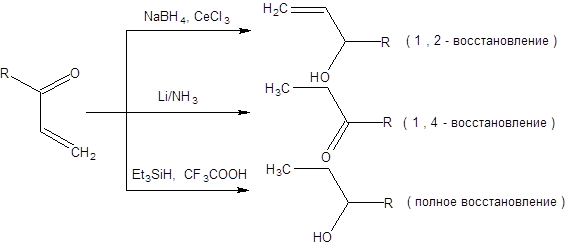Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Селективность органических реакцийСодержание книги
Поиск на нашем сайте
Планирование синтеза даже относительно простых соединений часто сопряжено с решением проблем селективности. Под селективностью понимают избирательность реакции по отношению к определенным группам атомов в молекуле или направлению подхода реагента. Надежность реакции как синтетического метода предполагает не только эффективное превращение данного структурного фрагмента в условиях реакции, но и требует гарантировать, что в условиях реакции протекает одна и только одна реакция по месту нахождения заданного структурного фрагмента. Проблема селективности столь многогранна, что однозначно определить ее границы весьма затруднительно, однако интуитивно вполне понятно, в каких случаях она возникает. Фундаментальное значение проблемы селективности при планировании и проведении многостадийных синтезов очевидно. Среди методов органического синтеза можно выделить селективные и специфичные методы. Селективные методы предполагают преимущественное протекание реакции по заданному структурному фрагменту. Специфические методы требуют полного протекания заданного превращения по заданному структурному фрагменту, таким образом: специфичность есть абсолютная селективность. Вместе с тем специфичность– категория в значительной степени абстрактная, так как нельзя достоверно утверждать, что превращение абсолютно избирательно. В настоящее время понятие специфичная реакция используется как синоним словосочетания «очень высокоселективная реакция». Различают несколько типов селективности органических реакций: 1. Хемоселективность –предпочтительное протекание реакции по одной из нескольких родственных, но химически различных функциональных групп. 2. Региоселективность –предпочтительное протекание реакции по определенному положению молекулы. 3. Стереоселективность – преимущественное образование в реакции определенного пространственного изомера. Стереоселективность реакции может быть двух типов:диастереоселективность и энантиоселективность. Если в ходе реакции создается только нужное стереохимическое отношение между структурными фрагментами (относительная конфигурация), то речь идет о диастереоселективных реакциях, т.е. реакциях селективных относительно образования определенных диастереомеров. Если реакция приводит к преимущественному образованию продукта определенной абсолютной конфигурации, то речь идет об энантиоселективности реакции, т.е. о преимущественном образовании одного из энантиомеров. Принципиальным является то, что энантиоселективность реакции достигается лишь при ее протекании через диастереомерные переходные состояния. Если проблему селективности рассмотреть с позиции кинетических особенностей протекающей реакции, то можно выделить три общих случая, обуславливающих проблему селективности [1]: 1. Протекание последовательных реакций. 2. Протекание параллельных реакций. 3. Протекание последовательно-параллельных реакций. Проблема селективности, связанная с протеканием последовательных реакций, возникает, когда возможно протекание последовательности превращений в условиях интересующей реакции. Например, проблема селективности, связанная с протеканием последовательных реакций возникает, если необходимо осуществить окисление первичного спирта до соответствующего альдегида. В общем случае при окислении первичного спирта возможно образование как альдегидов, так и карбоновых кислот. Последние образуются в результате дальнейшего окисления альдегидов в условиях реакции. В данном случае проблема селективности решается выбором подходящего окислителя. Известно, что диоксид марганца эффективно окисляет первичные спирты до альдегидов (схема 1.5). Напротив, использование бихромата калия в кислой среде приводит к преимущественному образованию карбоновой кислоты (схема 1.5).
Схема 1.5 Протекание последовательных реакций наблюдается также при восстановлении ацетиленов. Если стоит задача, получить алкен исходя из соответствующего алкина, без его дальнейшего восстановления до алкана, то задача, как и в случае получения альдегидов из первичных спиртов решается выбором подходящего реагента. Известно, что катализатор Линдлара селективно восстанавливает алкины в соответствующие алкены цис-конфигурации (Схема 1.6).
Схема 1.6
Проблема селективности, связанная с протеканием последовательных реакций, особенно характерна для процессов окисления и восстановления, однако ими не ограничивается. Так, например определенные затруднения, связанные с протеканием последовательных реакций, характерны в случае необходимости проведения моноалкилирования енолятов, при использовании в качестве алкилирующих реагентов алкилйодидов, а также аллил- и бензилгалогенидов. Образование продуктов диалкилирования карбонильных соединений, наряду с продуктами моноалкилирования, связано с возможностью вторичного депротонирования продукта моноалкилирования непрореагировавшим енолят-ионом, как показано на схеме 1.7.
Схема 1.7
В тех случаях, когда необходимо осуществить моноалкилирование карбонильных соединений, рекомендуется проводить реакцию при возможно более низкой температуре в неполярных апротонных растворителях, используя в качестве алкилирующих реагентов алкилбромиды или лучше алкилхлориды. Другая группа проблем обеспечения селективности связанна с возможностью протекания параллельных реакций в системе. Примером решения проблемы селективности, связанной с возможностью протекания нескольких параллельных реакций, может быть избирательное восстановление карбонильной группы в присутствии енонового фрагмента. В этом случае возможно селективное восстановление карбонильной функции под действием боргидрида натрия, т. е. селективность достигается выбором подходящего реагента (схема 1.8).
Схема 1.8
Часто проблема селективности связанна с возможностью протекания как последовательных, так и параллельных реакций в одной системе, например при алкилировании толуола в условиях реакции Фриделя – Крафтса. В этом случае возможно развитие следующей последовательности реакций, приводящей к образованию сложной смеси продуктов (схема 1.9):
Схема 1.9 Для селективного получения из толуола продуктов его алкилирования целесообразно проводить первоначальное ацилирование или формилирование, с последующим восстановлением карбонильной функции, так как карбонильная группа препятствует дальнейшему протеканию электрофильного замещения в бензойном кольце (схема 1.10).
Схема 1.10
Решение возникающих проблем селективности может быть осуществлено разными способами. К числу наиболее распространенных способов обеспечения селективности органических реакций относятся: 1. Выбор подходящей реакции. 2. Варьирование природы реагентов и условий проведения реакции. 3. Селективная активация альтернативных реакционных центров. 4. Использование защитных групп. Наиболее универсальным методом обеспечения необходимой селективности является использование защитных групп. Этот метод, однако, имеет очевидный недостаток, связанный с увеличением количества необходимых стадий для проведения заданной трансформации, так как использование защитных групп связано с необходимостью их первоначального введения и последующего удаления. Исходя из сказанного, очевидны требования к защитным группам: 1. Защитная группа должна легко ставиться (селективно, с выходом, близким к количественному). 2. Защитная группа должна быть инертна по отношению к реагентам, используемым для проведения трансформации других функциональных групп. 3. Защитная группа должна легко удаляться в мягких условиях. В настоящее время разработано значительное количество методов защиты функциональных групп, поэтому рассмотрим лишь наиболее широко используемые. А. Защита гидроксильной группы Защита спиртовой группы необходима при проведении реакций с участием других функциональных групп субстрата в сильноосновных средах (литийорганические соединения, реактивы Гриньяра, амиды щелочных металлов) или в присутствии сильных окислителей. Наибольшую популярность завоевала тетрагидропиранильная защита, которая легко вводится и легко снимается в кислой среде, оставаясь совершенно инертной по отношению к основаниям, а также сильным окислителям и восстановителям. Постановка и удаление тетрагидропиранильной защиты, а также пример ее использования приведены на схеме 1.11 [2].
Схема 1.11 Некоторое значение для защиты гидроксильной группы имеют бензильная и тритильная группы. Постановка этих защит осуществляется действием бензилбромида и тритилхлорида на спирты в присутствии третичных аминов, а удаление каталитическим гидрогенолизом, а в случае тритильной защиты также и кислотным расщеплением:
Схема 1.12
Также возможно использование метоксиметиловых эфиров в качестве защиты. Пример использования метоксиметиловых эфиров в качестве защиты приведен на схеме 2.13.
Схема 1.13 Для защиты спиртовой группы с успехом используется предложенный Кори йодметилтиометиловый эфир (схема 1.14).
Схема 1.14 Также интерес для маскирования гидроксильной группы представляет триметилсилильная и трет-бутилдиметилсилильная защиты, которые легко удаляются под действием фторид-иона в очень мягких условиях. Вместе с тем трет-бутилдиметилсилильная защита на четыре порядка более устойчива к гидролизу, чем триметилсилильная. Так триметилсильная защита снимается при действии воды уже в нейтральной среде, тогда как удаление трет-бутилдиметилсильной защитыосуществляется при рН <4. Применение трет-бутилдиметилсилильной защиты целесообразно при необходимости осуществления гидридного восстановления субстрата или окисления, однако ее неустойчивость по отношению к сильным основаниям значительно ограничивает возможности ее использования.
Схема 1.15 При выборе защиты гидроксильной группы следует учитывать специфику желаемого превращения и в первую очередь pH реакционной среды. Часто эффективность необходимой реорганизации структуры можно повысить, если использовать тандемную последовательность необходимой трансформации и удаления защиты. Б. Защита карбонильной группы Карбонильная функция – сильный электрофил, что во многом обуславливает заложенный в ней синтетический потенциал. Однаковысокая электрофильность карбонильной группы часто осложняет трансформации других функциональных групп под действием нуклеофильных агентов. В таких случаях необходимо понижать электрофильность карбонильной группы, что достигают ее превращением в ацетали или тиоацетали. Альдегиды и кетоны при взаимодействии со спиртами в присутствии сильных кислот превращаются в ацетали и кетали соответственно. Ацетали и кетали инертны к действию сильных оснований, а также по отношению к окислителям и восстановителям, однако лабильны в присутствии кислот, что позволяет легко удалять эти защитные группы в кислой среде. Наибольшее практическое значение имеет превращение карбонильной функции в производные 1,3-диоксолана и 1,3-диоксана взаимодействием с этиленгликолем или 1,3-пропиленгликолем в присутствии сильных кислот с азеотропной отгонкой воды. Примеры постановки ацетальной защиты и ее использования в органическом синтезе приведены на схеме 1.16.
Схема 1.16 Превращение карбонильной функции в соответствующие тиоацетали и тиокетали протекает в исключительно мягких условиях, равно как и удаление этой защитной группы (схема 1.17). Единственным ограничением является повышенная кислотность тиоацеталей и тиокеталей, что определяет их способность депротонироваться в присутствии таких сильных оснований, как литийорганические соединения и реактивы Гриньяра.
Схема 1.17
В. Защита карбоксильной группы Защита карбоксильной группы может преследовать две цели: 1. Устранение кислотных свойств, обусловленных подвижным протоном карбоксильной группы. 2. Понижение электрофильности углеродного атома, карбоксильной группы. Первая задача тривиальна и решается превращением карбоновых кислот в сложные эфиры. Наиболее часто используются метиловые и этиловые эфиры карбоновых кислот как наиболее доступные, однако это сопряжено с необходимостью применять достаточно жесткие условия для их гидролиза, а именно использование щелочей и повышенной температуры. Если необходимо избегать применения щелочей, то эффективными могут оказаться бензиловые эфиры, которые лабильны в условиях каталитического гидрогенолиза (схема.1.18).
Схема 1.18
Трет-бутиловые эфиры также нашли применение в качестве защитных групп, так как они легко гидролизуются даже в слабокислой среде. Некоторое применение нашли также 2,2,2-трихлорэтиловые эфиры карбоновых кислот, которые легко разрушаются под действием цинка в водной уксусной кислоте (схема 1.19).
Схема 1.19 Другой класс защитных групп обеспечивает понижение электрофильности углеродного атома карбоксильной функции и связан с ее трансформацией в менее электрофильную группу. Это достигается превращением карбоновых кислот в соответствующие 2 – оксазолиновые производные, как показано на схеме 1.20.
Схема 1.20
2-оксазолиновые производные не взаимодействуют с реактивами Гриньяра и гидридными восстановителями, что определяет их ценность как защиты для карбоксильной группы. Регенерация карбоксильной группы производится в условиях кислотного гидролиза 2-оксазолиновых производных карбоновых кислот. Г. Защита аминогруппы Присутствие аминогруппы в соединении в первую очередь обуславливает его нуклеофильность и основность, а также повышенную склонность к окислению, часто неконтролируемым образом. Понижение нуклеофильных свойств аминогруппы, а также способности к окислению можно достигнуть ее ацилированием. Например, известно, что прямое нитрование анилина невозможно, ввиду его окисления с образованием продуктов неустановленного строения. Однако синтез о- и п-нитроанилинов, исходя из анилина,– задача тривиальная, решающаяся превращением последнего в амиды, которые гладко нитруются. Последующее удаление ацетильной защиты щелочным гидролизом приводит к образованию нитроанилинов (схема 1.21).
Схема 1.21
Защита аминогруппы ацилированием ангидридами и хлорангидридами алифатических карбоновых кислот сопряжена с необходимостью последующего щелочного или кислотного гидролиза в достаточно жестких условиях. В существенно более мягких условиях удаляются бензилоксикарбонильная (Cbz) и трет-бутилоксикарбонильная (Boc) группы. Бензилоксикарбонильная группа вводится в исключительно мягких условиях, а удаляется гидрогенолизом на палладиевом катализаторе (схема 1.22).
Схема 1.22 Наибольшую популярность среди разнообразных защит аминогруппы приобрела трет-бутилоксикарбонильная группа, которая вводится, как бензилоксикарбонильная, в исключительно мягких условиях, а удаляется мягким кислотным гидролизом (схема 1.23).
Схема.1.23
Использование трет-бутилоксикарбонильной защиты направлено на понижение нуклеофильности аминогруппы, что зачастую бывает необходимо при проведении ряда синтезов. Например, один из вариантов реализации твердофазного синтеза пептидов по Меррифилду сопряжен с использованием Вос-защиты аминогруппы исходных аминокислот. В синтезе Меррифилда в качестве твердофазного носителя используется хлорметилированный трехмерный сополимер стирола и п-дивинилбензола, получаемый как показано на схеме 1.24:
Схема 1.24 Хлорметильные группы, введенные в полимерный носитель, далее взаимодействуют с аминокислотами, защищенными по аминогруппе исключительно по карбоксильной группе в присутствии третичного амина. Таким образом, осуществляется иммобилизация С-концевой аминокислоты синтезируемого пептида. Полученный продукт демаскируют в кислой среде и далее вводят следующую защищенную по аминогруппе аминокислоту. Введение в реакционную систему дициклогексилкарбодиимида (DСС) приводит к образованию пептидной связи между уже иммобилизованной и вновь введенной аминокислотой. Повторение рассмотренных операций заданное число раз приводит к синтезу полипептидов заданного строения (схема 1.25):
Схема 1.25 По окончании формирования заданной аминокислотной последовательности полипептидную цепь освобождают от носителя мягким кислотным гидролизом смесью бромоводородной и трифторуксусной кислот (схема 1.26).
Схема 1.26
Основным достоинством метода Меррифилда является его твердофазность, обеспечивающая легкость отделения продуктов реакции от исходных веществ обыкновенным фильтрованием, а также возможность автоматизации процесса. Метод Меррифилда, однако, не лишен присущих ему недостатков, главным из которых является линейных характер построения синтетической схемы так, что даже выход в 99,8% на каждой стадии, при значительной длине требуемой аминокислотной последовательности, приводит к «проскокам» аминокислот. За разработку твердофазного синтеза пептидов Меррифилду была присуждена Нобелевская премия 1984 года. Использование защитных групп является наиболее универсальным методом контроля селективности, однако часто ее можно достичь и другими средствами, которые подчас дают лучшие результаты. Так хемоселективность можно контролировать просто выбором подходящей реакции. Например, восстановление изолированных диенов с двойными связями разной степени замещения может быть успешно проведено по требуемой кратной связи просто изменением условий восстановления (схема 2.2.27).
Схема 2.2.27
В случае гетерогенного каталитического гидрирования восстановлению подвергается наименее замещенная двойная связь, так как ход процесса контролируется преимущественно стерическими факторами. Гидрирование в кислой среде в присутствии источника гидрид-иона, напротив, контролируется электронными факторами, а именно, первоначальным образованием наиболее устойчивого карбкатиона, который далее отщепляет гидрид-ион от триэтилсилана, с образованием продукта гидрирования по наиболее замещенной двойной связи (схема 2.2.27). Иной подход к обеспечению селективности заключается в выборе подходящего реагента. Выбор подходящего реагента – удобный и в значительной степени универсальный путь решения проблем селективности. Выбором подходящего реагента можно достичь не только хемоселективности, но также регио- и стереоселективности. Например, выбором подходящего реагента возможно решить проблему селективного замещения первичной гидроксильной группы в присутствии вторичной гидроксильной группы (схема 1.28). Под действием квазифосфониевых солей, скорость взаимодействия которых со спиртами убывает в ряду: первичные >вторичные > третичные, возможно заместить на галоген первичную гидроксильную группу в присутствии вторичного гидроксила. Еще большую селективность замещения первичной гидроксильной группы на галоген обеспечивают системы Ph3P + CCl4 и Ph3P + NBS. Последняя система столь эффективна для решения поставленной задачи, что нашла применение вхимии углеводов (схема 1.28):
Схема 1.28
Классическим примером решения проблем селективности выбором подходящего реагента является восстановление α,β-ненасыщенных карбонильных соединений. В зависимости от природы восстановителя можно добиться селективного восстановления по одному из трех альтернативных направлений (схема 1.29):
Схема 1.29
Еще одним методом контроля селективности является избирательная активация альтернативных реакционных центров субстрата. В качестве примера активации альтернативных реакционных центров рассмотрим алкилирование 2-бромпропенола-2. При действии одного эквивалента н-бутиллития на субстрат депротонируется гидроксильная группа как наиболее кислая, поэтому дальнейшее алкилирование возможно только по атому кислорода. При действии двух эквивалентов н-бутиллития образуются два альтернативных нуклеофильных центра, однако нуклеофильность атома углерода существенно выше, что предопределяет преимущественное алкилирование по атому углерода. Таким образом, в первом случае наблюдалась селективная активация гидроксильной группы, а во втором – бромида винильного типа (схема 1.30).
Схема 1.30 Список литературы 1. Смит В. А., Бочков А. Ф., Кейпл Р. Органический синтез. М.: Мир, 2001. С. 131 – 195. 2. Кери Ф., Сандберг Р. Углубленный курс органической химии. М.: Химия, 1981. С. 356 – 364.
|
||
|
|
Последнее изменение этой страницы: 2021-05-27; просмотров: 1369; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.137 (0.014 с.) |