
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Теория переходного состоянияСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Цель, которую ставили перед собой исследователи, создавшие эту теорию (Эйринг, Винн-Джонс, Ледлер, Поляни), заключалась в разработке способа, позволяющего вычислять абсолютную скорость реакции из данных, характеризующих молекулы или атомы реагирующих веществ. Поэтому теорию переходного состояния или переходного комплекса часто называют теорией абсолютных скоростей. Цель была достигнута не полностью — пришлось сделать ряд произвольных допущений. Теория приобрела полуэмпирический характер, но тем не менее основные ее идеи оказались очень плодотворными и стимулировали развитие современной кинетики. Теория постулирует, что реагирующие молекулы сначала образуют переходный «активированный комплекс», находящийся в равновесии с исходными веществами. Затем этот комплекс превращается в конечные продукты, причем скорость реакции определяется именно скоростью разложения переходного комплекса X: А+В→[X]→C+D где X — переходный комплекс, часто обозначаемый звездочкой: [Х]=[АВ]*. Комплекс сходен с обычной молекулой, отличие состоит в том, что он непрерывно изменяется, приближаясь к конфигурации конечных продуктов. Эйринг показал, что между константой скорости реакции и константой равновесия комплекса существует соотношение
где h — постоянная Планка; N — постоянная Авогадро; ϰ — трансмиссионный коэффициент. Применяя к константе К* обычные термодинамические правила, получим зависимость
Сравнивая эти два уравнения, найдем основное уравнение теории абсолютных скоростей
Все величины, отмеченные звездочкой, относятся к переходному комплексу. Величина ∆H* определяется из найденной опытным путем энергии активации Е: для реакций первого порядка ∆H*=E—RT, для реакции второго порядка ∆H*=E—2RT. Теоретическое вычисление ∆S* представляет большие трудности, но общий вид зависимости скорости от ∆S* и ∆H* ясен. Реакция будет протекать тем быстрее, чем больше фактор e∆S*/R т. е. чем больше энтропия активации. Большое значение ∆S* означает, что переходное состояние мало упорядочено, переходный комплекс обладает «рыхлой» структурой. Возможно, что реакция будет протекать быстро, несмотря на большую энергию активации (такой случай усматривали в процессе денатурации белков, протекающем быстро, хотя энергия активации значительна). При небольшом изменении энтропии скорость определяется другим конкурирующим фактором, именно ∆H*. Здесь возрастание ∆H* соответствует уменьшению скорости. Вопрос 24. Химические реакции, протекающие на границе раздела двух фаз, называют гетерогенными. Гетерогенные процессы широко распространены в природе и часто используются в практике. Примерами могут служить процессы растворения, кристаллизации, испарения, конденсации, химические реакции на границе раздела двух фаз, гетерогенный катализ и др.
знак минус ставят, когда скорость выражается через убыль концентрации исходного вещества, чтобы скорость была положительной величиной. Скорость выражается через прирост концентрации иода водорода, разделана на стехиометрический коэффициент 2, чтобы все три выражения были численно одинаковы. Из вышеприведённых формул следует, что размерность скорости химической реакции равна: химическая масса * объём -1 * время -1. В системе СИ размерность скорости равна - моль * м -3 * сек -1. Однако в литературе до сих пор принято выражать скорость реакций в моль * дм -3 * сек -1. На практике при измерении скоростей реакции вместо секунды за единицу времени часто берут минуту или час. В гетерогенных системах реакционным пространством является поверхность раздела между фазами. Введя понятие поверхностной концентрации Cs, измеряемой числом молей реагирующего вещества (n), приходящихся на единицу реакционного пространства S, имеем Лимитирующая стадия процесса. При стационарном процессе, когда все стадии протекают с одинаковой скоростью, одна из них — наиболее медленная — имеет максимальную в данных условиях скорость. В зависимости оттого, какая из стадий ограничивает скорость протекания процесса, различают диффузионную и электрохимическую кинетику. При изучении процесса важно выявить, какая из стадий ограничивает его скорость. Тагё^ например, если процесс протекает с концентрационной поляризацией, то эффективной мерой, позволяющей ускорить процесс, является перемешивание. Если же лимитирующей является химйческая поляризация, то перемешивание почти не сказывается на скорости процесса. В последнем случае повышение температуры способствует росту скорости реакции. Для определения лимитирующей стадии электродного процесса разработаны методы температурной зависимости дифференциальной полярографии [68] и метод быстрого снятия поляризационных кривых [9]. Вопрос 25. Гетерогенный катализ Если катализаторы и реагенты находятся в разных фазах и имеют границу раздела, то катализ называется гетерогенным. Катализатор является твердым веществом, а реагирующие вещества – газы или жидкости. Реагирующие молекулы адсорбируются на поверхности катализатора, и за счет ориентации определенным образом и ослабления внутримолекулярных связей снижается энергия активации и увеличивается скорость реакции. Пусть в отсутствии катализатора протекает реакция А + В = АВ* = Продукты, а в присутствии катализатора скорость ее возрастает, но продукты остаются теми же. Если считать, что активное адсорбционное состояние аналогично активированному комплексу АВ некаталитической реакции, то весь процесс можно изобразить следующим образом. 1. Адсорбция исходных веществ на поверхность катализатора: А + В + Кт = АВКт Как правило, этот процесс экзотермический. 2. Перевод адсорбированного состояния в активное: АВКт = АВКт* Этот процесс требует затраты энергии, называемой истинной энергией активации. 3. Реакция в адсорбированном состоянии с образованием адсорбированных конечных продуктов: АВКт* = Продукты Кт 4. Десорбция продуктов реакции, приводящая к регенерации катализатора: Продукты Кт = Продукты + Кт Таким образом, и в гетерогенном катализе ускоряющее действие катализатора, так же как и в гомогенном катализе, связано с тем, что реагирующие вещества образуют промежуточные соединения, что приводит к снижению энергии активации. КАТАЛИЗ - процесс, заключающийся в изменении скорости химических реакций в присутствии веществ, называемых катализаторами. Катализаторы - вещества, изменяющие скорость химической реакции, которые могут участвовать в реакции, входить в состав промежуточных продуктов, но не входят в состав конечных продуктов реакции и после окончания реакции остаются неизменными. Каталитические реакции - реакции, протекающие в присутствии катализаторов. Положительным называют катализ, при котором скоость реакции возрастает, отрицательным (ингибированием) - при котором она убывает. Примером положительного катализа может служить процесс окисления аммиака на платине при получении азотной кислоты. Примером отрицательного - снижение скорости коррозии при введении в жидкость, в которой эксплуатируется металл, нитрита натрия, хромата и дихромата калия. Катализаторы, замедляющие химическую реакцию, называются ингибиторами. В зависимости от того, находится катализатор в той же фазе, что и реагирующие вещества, или образует самостоятельную фазу, говорят о гомогенном или гетерогенном катализе. Примером гомогенного катализа является разложение пероксида водорода в присутствии ионов йода. Реакция протекает в две стадии: Н О + I = H O + IO Н O + IO = Н O + O + I При гомогенном катализе действие катализатора связано с тем, что он вступает во взаимодействие с реагирующими веществами с образованием промежуточных соединений, это приводит к снижению энергии активации. При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела - катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель. Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы. 1. Диффузия реагирующих веществ к поверхности твердого вещества. 2. Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их. 3. Химическая реакция между реагирующими молекулами. 4. Десорбция продуктов с поверхности катализатора. 5. Диффузия продукта с поверхности катализатора в общий поток. Примером гетерогенного катализа является окисление SO в SO на катализаторе V O при производстве серной кислоты (контактный метод). Промоторы (или активаторы) - вещества, повышающие активность катализатора. При этом промоторы могут сами и не обладать каталитическими свойствами. Каталитические яды - посторонние примеси в реакционной смеси, приводящие к частичной или полной потере активности катализатора. Так, следы мышьяка, фосфора вызывают быструю потерю катализатором V O активности (контактный метод производства H SO). Все Опыт показывает, что при гетерогенном катализе большое значение имеют структура, химический состав и величина поверхности катализатора. Так, гладкая платиновая пластинка, опущенная в раствор пероксида водорода, практически не вызывают разложения последнего. Если же поверхность этой пластинки шероховатая, разложение пероксида водорода протекает заметно. Ещё больше увеличивается скорость разложения, если в качестве катализатора использовать платину в виде порошка. И, наконец, если к раствору пероксида водорода прибавить еще более раздробленную платину в виде её коллоидного раствора, то разложение Н2О2 произойдёт практически мгновенно - в виде взрыва. Как правило все гетерогенные каталитические реакции (даже относительно простые из них) протекают в несколько стадий: 1) сближение молекул реагирующих веществ, 2) ориентация молекул на активных центрах катализатора, 3) адсорбция молекул реагирующих веществ, сопровождающаяся деформацией связей в молекулах, 4) химическое превращение адсорбированных (и активированных) молекул, 5) десорбция продуктов реакции, 6) удаление этих продуктов с поверхности катализатора.
ГЕТЕРОГЕННЫЙ КАТАЛИЗ (контактный катализ), изменение скорости хим. р-ции при воздействии катализаторов, образующих самостоят. фазу и отделенных от реагирующих в-в границей раздела. наиб. распространен случай, когда твердый кат. (контакт) ускоряет р-цию между газообразными реагентами или р-цию в р-ре. Каталитич. р-ция протекает обычно на пов-сти твердого кат. и обусловлена активацией молекул реагентов при взаимод. с пов-стью. Поэтому для осуществления Г. к. необходима адсорбция компонентов реакц. смеси из объемной фазы на пов-сти катализатора. В техн. Г. к. св-во катализатора ускорять р-цию обычно определяют как выход продукта в единицу времени, отнесенный к единице объема или массы катализатора. В теоретич. исследованиях скорость v гетерогенно-каталитич. р-ций относят к единице пов-сти катализатора и наз. удельной каталитич. активностью; ее размерность - моль х х с-1м-2(см. Активность катализатора). Если все активные центры пов-сти однородны и равнодоступны молекулам реагирующих в-в, v пропорциональна пов-сти S:
где f- ф-ция концентраций сA, сB,... реагентов и продуктов р-ции в объемной фазе, k -константа скорости, отнесенная к единице пов-сти катализатора. Соотношение (1) м. б. неприменимо, если гетерогенно-каталитич. р-ция осложнена диффузией реагирующих молекул, в случае неоднородной пов-сти и др. (подробнее см. Каталитических реакций кинетика). Развитие эксперим. техники сделало возможным в ряде случаев относить скорость гетерогенно-каталитич. р-ции к единичному активному центру пов-сти. Применение сверхвысокого вакуума (~10-8Па) позволило получить атомарно чистые пов-сти металлов (свободные от адсорбиров. частиц), на к-рых все атомы (их число Nc ~ 1019 м-2) являются активными центрами. Число молекул, подвергающихся превращениям на одном активном центре в секунду, наз. числом оборотов р-ции tn. Скорость р-ции связана с tn соотношением:
Для многих р-ций tn составляет от 10-2 до 102 с -1 однако возможны как существенно меньшие, так и большие значения. Напр., в случае окисления NH3 на пром. катализаторе tn достигает 5*104 с-1, а для той же р-ции на монокристалле Pt при низком давлении -10-6 с-1. Число активных центров на катализаторах-оксидах металлов оценить труднее. Спектральные методы (оптическая и ИК-спектроскопия, ЭПР и др.) позволяют следить за изменением структуры пов-сти этих в-в в условиях Г. к. и соотносить эти изменения с изменениями скорости р-ции; отсюда можно оценить поверхностную концентрацию активных центров. Оказалось, что для большинства оксидных кат. число активных центров меньше, чем для металлов, и составляет 1014-1018 м-2. Константы скорости каталитич. р-ции изменяются с т-рой Г в соответствии с ур-нием Аррениуса: k = = k0ехр(— E/RT), где k0- предэкспоненц. множитель, Е- энергия активации, R- газовая постоянная. Величина k0 м. б. вычислена методами активированного комплекса теории, однако сопоставление расчетных значений с экспериментальными возможно лишь при условии, если точно известно число активных центров на пов-сти реального катализатора. Значения Е определяются из эксперим. зависимости k от Т, обычно они составляют 10-150 кДж/моль. Катализаторы Г. к. обладают высокой селективностью, т. е. относит. способностью ускорять одну из неск. одновременно протекающих р-ций. Часто на одном и том же катализаторе могут протекать одновременно неск. последовательных и параллельных р-ций. Напр., каталитич. окисление углеводородов может протекать последовательно-сначала до получения ценных кислородсодержащих соед., затем до полного их окисления с образованием СО2 и Н2О. Наряду с этим может протекать и параллельное окисление исходного в-ва непосредственно до СО2 и Н2О, без выхода ценных промежут. продуктов мягкого окисления в газовую фазу. В простейшем случае мягкого окисления углеводорода СnНm+2, до продукта СnНmО (напр., при получении этиленоксида С2Н2О из этилена С2Н4) процесс м. б. описан схематически след, образом: Селективность s катализатора определяется как отношение скорости накопления целевого продукта к сумме скоростей всех р-ций, протекающих с участием исходных в-в: Стадия гетерогенного катализа -Сложная гетерогенно-каталитич. р-ция протекает через последовательность стадий, традиционно считавшихся элементарными: подвод реагентов из объемной фазы к пов-сти катализатора (диффузия), адсорбция, хим. превращение на пов-сти (собственно катализ), десорбция продуктов, их отвод от пов-сти катализатора (диффузия). Исходными в-вами, реагирующими в элементарных р-циях, по определению, являются промежут. в-ва, к-рые образуются в предшествующих стадиях и не м. б. выделены вместе с реагирующими в-вами или продуктами р-ции. Предполагалось, что в Г. к. такими в-вами являются поверхностные соед. катализатора с реагентами, а стадии, в к-рых эти соед. образуются или распадаются,-элементарные. Развитие эксперим. методов исследования позволило установить, что в нек-рых из стадий, предполагавшихся ранее элементарными, можно выделить свои элементарные процессы, в к-рых промежут. в-ва не образуются, а изменение состояния частицы (атома, молекулы) связано с преодолением одного потенциального барьера. Рассмотрим влияние каждой из стадий на кинетич. закономерности Г. к. Диффузия. Если катализатор представляет собой пористые частицы (зерна), в общем случае могут иметь место след. диффузионные процессы: 1) перенос реагирующих в-в из объема газовой или жидкой фазы к внеш. пов-сти зерна катализатора (внешняя диффузия); 2) перенос частиц в порах внутри зерна (внутренняя диффузия); 3) и 4) обратные процессы - перенос частиц продуктов р-ции изнутри пор к внеш. пов-сти зерен и отвод их от внеш. пов-сти зерен в пространство между ними (соотв. внутр. и внеш. диффузия). Все эти процессы обусловлены градиентом концентраций компонентов реакц. смеси в пределах одной фазы или у границы раздела фаз. В зависимости от условий осуществления процесса (т-ры, давления, св-в катализатора) различают кинетич. область протекания Г. к., в к-рой все диффузионные процессы происходят значительно быстрее, чем хим. р-ции, и диффузионные области (соотв. внешне- и внутренне-диффузионную), в к-рых суммарный процесс тормозится более медленным, чем хим. р-ция, переносом частиц. Поскольку с ростом т-ры и концентрации реагентов скорость хим. р-ции увеличивается быстрее, чем диффузия, диффузионное торможение в Г. к. происходит при высоких т-рах и давлениях, а также на высокодисперсных катализаторах. Если ур-ние р-ции в кинетич. области имеет вид:
где n-порядок р-ции, с-концентрация реагирующего в-ва в объемной фазе, то с повышением т-ры и переходом во внутренне-диффузионную область скорость р-ции описывается ур-нием: где S*-внеш. пов-сть зерна катализатора, D*- эффективный коэф. диффузии в пористой среде. Поскольку в ур-ние входит константа скорости k в степени 1/2, измеряемая энергия активации р-ции во внутренне-диффузионной области уменьшается вдвое по сравнению с ее значением в кинетич. области, а порядок р-ции по реагенту, диффузия к-рого лимитирует суммарную скорость процесса, изменяется до значения (п + 1)/2. При еще более высоких т-рах, когда процесс переходит во внешне-диффузионную область, температурная зависимость кажущейся константы скорости определяется температурной зависимостью коэф. диффузии (см. рис. 1). Подробнее см. в ст. Макрокинетика. Рис. 1. Зависимость константы скорости k гетерогенно-каталитич. р-ции от т-ры Т в кинетической (1),внутренне-диффузионной (2) и внешне-диффузионной (3) областях протекания. Адсорбция. Согласно общепринятым представлениям, основное значение в Г. к. имеет хемосорбция, при к-рой адсорбируемые частицы химически связаны с поверхностными атомами твердого тела. Для участия в послед. каталитич. превращениях хемосорбиров. частица (атом, молекула) должна быть активирована, т. е. переведена в более реакционноспособное (по сравнению с исходным) состояние. Этот процесс может требовать затраты энергии (преодоления энергетич. барьера), протекать медленно и оказаться лимитирующей стадией Г. к. Часто медленную хемосорбцию в Г. к. наз. активированной адсорбцией. Примером процесса, лимитируемого активиров. адсорбцией, является синтез NH3 на железном кат., скорость к-рого определяется адсорбцией N2 на пов-сти Fe. Энергия связи хемосорбиров. частицы с пов-стью катализатора не должна быть ни слишком большой, ни слишком малой. Так, изменение скорости окисления водорода на пов-сти разл. оксидов металлов имеет максимум, соответствующий оптимальной для катализа теплоте адсорбции Qs кислорода (рис. 2). Слабая связь (напр., при физ. адсорбции) не приводит к активации адсорбиров. частицы и образованию более реакционноспособного состояния, а слишком прочная связь затрудняет дальнейшие превращ. (повышает энергию активации послед. стадии). Причины активиров. адсорбции при Г. к. могут быть различными. Больших энергетич. затрат может требовать перестройка поверхностной структуры катализатора. Напр., при адсорбции Н2 или СО на грани (100) кубич. монокристаяла Pt гексагональная структура перестраивается в квадратную. Элементарными процессами активации м. б. также перенос электрона от катализатора к хемосорбиров. молекулам с образованием ионов или своб. радикалов, подвод энергии к адсорбиров. частице с образованием колебательно- или электронно-возбужденных молекул, взаимная ориентация атомов или атомных групп хемосорбиров. молекул, благоприятная для послед. образования реакционноспособных комплексов, напр. Рис. 2. Зависимость константы скорости k окисления Н2 от теплоты адсорбции Q, кислорода (энергии связи металл -кислород). Во мн. процессах Г. к. адсорбция реагирующих в-в на пов-сти катализатора происходит через образование т. наз. предсорбционного состояния, или прекурсора, к-рое далее либо участвует в катализе, либо препятствует ему. Так, при окислении СО на МnО2 образуется предсорбц. состояние МnО2(СО), к-рое далее может участвовать в образовании продукта СО2 либо ведет к прочной хемосорбции с образованием поверхностного карбоната МnСО3, к-рый отравляет катализатор: Др. пример-образование на пов-сти слабо связанного прекурсора О*2, диффундирующего к разл. активным центрам, на к-рых он может либо перейти в прочно адсорбированный атомарный кислород, либо взаимодействовать, напр., с молекулой NH3, адсорбированной на Pt: Установлено, что число мест на пов-сти для адсорбции О2 в виде прекурсора значительно больше числа активных центров каталитич. превращения. Предполагается, что прекурсором м. б. колебательно- или электронно-возбужденная молекула. Каталитическое превращение на поверхности. Считается, что катализ протекает по двум основным механизмам. В случае механизма Ленгмюра-Хиншельвуда в р-ции участвуют только адсорбиров. частицы, а скорость р-ции пропорциональна заполнениям
где где РА-парциальное давление реагента А. Однако установлено, что прочно адсорбиров. молекулы могут непосредственно взаимод. между собой, лишь если они занимают соседние активные центры (соседние атомы пов-сти катализатора), что в большинстве случаев маловероятно. Как правило, для Г. к. необходимо, чтобы одна из частиц (напр., А) перешла в состояние слабой адсорбции и продиффундировала ко второй частице (В). Элементарной стадией катализа при этом может оказаться именно поверхностная диффузия. Исследования методом молекулярных пучков показали, что ударный механизм Г. к. в чистом виде практически не наблюдается. При впуске молекул А на катализатор с адсорбированными на нем молекулами В (напр., при впуске Н2 на Pt, покрытую D2) продукт (HD) в отраженном пучке появляется не сразу, а спустя нек-рое время, необходимое для диффузии молекул А к активным центрам катализатора. Ур-ние (7) в этом случае соблюдается. Десорбция. Простая десорбция происходит путем разрыва связи молекулы продукта с пов-стью. Прочность хим. связи составляет 200-400 кДж/моль и существенно превышает энергию активации каталитич. р-ции. В условиях Г. к. часто происходит не простая десорбция, а десорбция через ассоциативный комплекс, почти не требующая затрат энергии. Напр., гидроксильная группа на пов-сти А12О3 переходит в газовую фазу при взаимод. с водяным паром или спиртом по бимолекулярному механизму:
Если группа OR пов-сти разлагается далее на группу ОН и молекулу олефина, протекает каталитич. дегидратация спирта.
|
||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 739; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.214 (0.017 с.) |

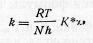









 аллильных комплексов при адсорбции олефинов, и др.
аллильных комплексов при адсорбции олефинов, и др. 


 пов-сти (долям пов-сти, занятым адсорбиров. частицами). Для р-ции типа А + В -> продукты скорость превращения выражается соотношением:
пов-сти (долям пов-сти, занятым адсорбиров. частицами). Для р-ции типа А + В -> продукты скорость превращения выражается соотношением:
 и
и  -заполнения пов-сти молекулами А и В соотв., определяемые в случае однородной пов-сти и обратимой адсорбции изотермой Ленгмгора (см. Адсорбция). При т. наз. ударном механизме (механизме Ридила-Или) частица А из газовой фазы сталкивается с адсорбированной на пов-сти частицей В, образуя продукты р-ции. В этом случае
-заполнения пов-сти молекулами А и В соотв., определяемые в случае однородной пов-сти и обратимой адсорбции изотермой Ленгмгора (см. Адсорбция). При т. наз. ударном механизме (механизме Ридила-Или) частица А из газовой фазы сталкивается с адсорбированной на пов-сти частицей В, образуя продукты р-ции. В этом случае 




