
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Разветвленные цепные реакции.Содержание книги
Поиск на нашем сайте
Термин предложен Н. Н. Семеновым для открытых им (1926-28) цепных реакций с критич. явлениями, состоящими в том, что незначит. измененияконцентрации реагентов, т-ры, размеров сосуда, введение примеси (даже разбавление реакционной смеси инертным газом) могут приводить к скачкообразному росту скорости цепных реакций от практически ненаблюдаемой до столь большой, что ее невозможно измерить. Впервые критич. явления в хим. системах были обнаружены Семеновым, Ю. Б. Харитоном и 3. Ф. Вальта при окислении паров фосфора. Термином "разветвление" обозначают входящую в такие р-ции стадию размножения активных частиц.
где п = wi /(g -f). Если же f станет больше g, т. е.
Выражение для скорости цепных реакций, известное как ур-ние Семенова, имеет вид:
т. е. концентрация активных частиц не принимает стационарного значения, а непрерывно нарастает, как и пропорциональная ей скорость цепнойреакции, переходящей в самовоспламенение. Условие g = f, или
Рис. 1. Динамика разветвленного цепного процесса при различных соотношениях факторов обрыва и разветвления цепи (соотв. g и f, Переход от стационарного к нестационарному протеканию цепной реакции может происходить как при увеличении f, так и при уменьшении g. Очевидно, что чем ближе условия к граничному f = g, тем меньшие изменения f или g могут привести к срыву и переходу р-ции в режим самоускорения.
Суммарная р-ция:
т. е. в ЦПЦ не только образуется продукт Н2О и регенерируются переносчики цепи
Все известные разветвленно-цепные р-ции являются экзотермическими, причем часть выделяемой энергии переходит в энергию активных частиц. Поэтому для разветвления цепи необходимо, чтобы тепловой эффект брутто-процесса значительно превосходил тепловой эффект наиб. энергоемкой из всех стадий разветвления (для окисления Н2 это соотв. 483 и 70 кДж/моль).
Рис. 2. Полуостров воспламенения смеси водорода с кислородом. Самоускорение цепных реакций с разветвлением цепей, описываемое ур-нием Семенова, обусловлено переходом энергии экзотермич. р-ции в хим. энергию активных частиц. Так, в определенных условиях до половины кол-ва молекул Н2 в режиме самовоспламенения может существовать в формеатомов Н2 + F2 Аналогичная р-ция Н2 + С12
Для аналогичной р-ции с участием С12 необходимо затратить 248 кДж/моль, т. е. такая р-ция практически не происходит. Р-ции продолжения цепи для обеих систем подобны: Энергия, выделяющаяся в р-циях 7,2 и 2', частично переходит в энергию колебат. возбуждения продуктов - соотв. HF и НС1. При последующих соударениях избыточная колебат. энергия рассеивается - переходит в поступат. и вращат. энергию мн. других частиц.
Энергии колебат. возбуждения близки лишь для пары HF-H2. Между этими частицами может протекать реакция
Сумарная р-ция:
Все особенности разветвленно-цепных р-ций - существование нижнего и верхнего предельных давлений, наличие полуострова воспламенения, зависимость положения нижнего предела от размеров сосуда и т. п.- присущи и этой р-ции с энергетич. разветвлением цепи.
Суммарная р-ция:
По механизму цепных реакций с энергетич. разветвлением происходят газофазное фторирование СН4 и его галогенпроизводных, фторированиеэтана, окисление тетрафторэтилена и ряда хлорзамещенных олефинов, хлорирование силана и нек-рые др. процессы. Поскольку в цепных реакций с энергетич. разветвлением возникает инверсная заселенность колебат. уровней, такие цепные реакции представляют практич. интерес для решения проблемы создания лазеров с хим. накачкой (см. Лазеры химические). Р-ции с вырожденным разветвлением. Такое назв. получили многочисленные радикально-цепные р-ции, для к-рых характерно самоускорение, описываемое ур-нием Семенова с очень малыми значениями
Рис. 3. Динамика процессов с вырожденным разветвлением цепей (1) и разветвленного цепного (2). Заштрихованные площади отражают кол-вореагентов, прореагировавших к моменту времени t. Рассмотрим, напр., цепное окисление углеводородов. В этом случае ЦПЦ включает след. р-ции:
В отсутствие инициатора инициирование происходит по р-ции:
Радикалы
Последующее быстрое образование переносчика цепи
Т. обр., по мере протекания р-ции и накопления ROOH скорость инициирования растет:
Чем больше wi, тем больше скорость образования ROOH, a чем больше [ROOH], тем сильнее ускоряется инициирование. Так реализуется положит. обратная связь в случае р-ций с вырожденным разветвлением цепей. Суммарная р-ция:
Это ур-ние ничем не отличается от аналогичного для разветвленно-цепных р-ций, и если бы распад ROOH на
Поскольку Вопрос28. Теория электролитической диссоциации (С. Аррениус, 1887г.)
1. При растворении в воде (или расплавлении) электролиты распадаются на положительно и отрицательно заряженные ионы (подвергаются электролитической диссоциации). 2. Под действием электрического тока катионы (+) двигаются к катоду (-), а анионы (-) – к аноду (+). 3. Электролитическая диссоциация - процесс обратимый (обратная реакция называется моляризацией). 4. Степень электролитической диссоциации (a) зависит от природы электролита и растворителя, температуры и концентрации. Она показывает отношение числа молекул, распавшихся на ионы (n) к общему числу молекул, введенных в раствор (N).
a = n / N 0<a<1
Механизм электролитической диссоциации ионных веществ
При растворении соединений с ионными связями (например, NaCl) процесс гидратации начинается с ориентации диполей воды вокруг всех выступов и граней кристаллов соли. Ориентируясь вокруг ионов кристаллической решетки, молекулы воды образуют с ними либо водородные, либо донорно-акцепторные связи. При этом процессе выделяется большое количество энергии, которая называется энергией гидратации. Энергия гидратации, величина которой сравнима с энергией кристаллической решетки, идет на разрушение кристаллической решетки. При этом гидратированные ионы слой за слоем переходят в растворитель и, перемешиваясь с его молекулами, образуют раствор.
Механизм электролитической диссоциации полярных веществ
Аналогично диссоциируют и вещества, молекулы которых образованы по типу полярной ковалентной связи (полярные молекулы). Вокруг каждой полярной молекулы вещества (например, HCl), определенным образом ориентируются диполи воды. В результате взаимодействия с диполями воды полярная молекула еще больше поляризуется и превращается в ионную, далее уже легко образуются свободные гидратированные ионы.
Электролиты и неэлектролиты
Электролитическая диссоциация веществ, идущая с образованием свободных ионов объясняет электрическую проводимость растворов. Процесс электролитической диссоциации принято записывать в виде схемы, не раскрывая его механизма и опуская растворитель (H2O), хотя он является основным участником.
CaCl2 «Ca2+ + 2Cl- KAl(SO4)2 «K+ + Al3+ + 2SO42- HNO3 «H+ + NO3- Ba(OH)2 «Ba2+ + 2OH-
Из электронейтральности молекул вытекает, что суммарный заряд катионов и анионов должен быть равен нулю. Например, для Al2(SO4)3 –– 2 • (+3) + 3 • (-2) = +6 - 6 = 0 KCr(SO4)2 –– 1 • (+1) + 3 • (+3) + 2 • (-2) = +1 + 3 - 4 = 0
Сильные электролиты
Это вещества, которые при растворении в воде практически полностью распадаются на ионы. Как правило, к сильным электролитам относятся вещества с ионными или сильно полярными связями: все хорошо растворимые соли, сильные кислоты (HCl, HBr, HI, HClO4, H2SO4,HNO3) и сильные основания (LiOH, NaOH, KOH, RbOH, CsOH,Ba(OH)2,Sr(OH)2,Ca(OH)2). В растворе сильного электролита растворённое вещество находится в основном в виде ионов (катионов и анионов); недиссоциированные молекулы практически отсутствуют.
Слабые электролиты
Вещества, частично диссоциирующие на ионы. Растворы слабых электролитов наряду с ионами содержат недиссоциированные молекулы. Слабые электролиты не могут дать большой концентрации ионов в растворе.
К слабым электролитам относятся: 1) почти все органические кислоты (CH3COOH, C2H5COOH и др.); 2) некоторые неорганические кислоты (H2CO3, H2S и др.); 3) почти все малорастворимые в воде соли, основания и гидроксид аммония (Ca3(PO4)2; Cu(OH)2; Al(OH)3; NH4OH); 4) вода. Они плохо (или почти не проводят) электрический ток. СH3COOH «CH3COO- + H+ Cu(OH)2 «[CuOH]+ + OH- (первая ступень) [CuOH]+ «Cu2+ + OH- (вторая ступень) H2CO3 «H+ + HCO- (первая ступень) HCO3- «H+ + CO32- (вторая ступень)
Неэлектролиты
Вещества, водные растворы и расплавы которых не проводят электрический ток. Они содержат ковалентные неполярные или малополярные связи, которые не распадаются на ионы. Электрический ток не проводят газы, твердые вещества (неметаллы), органические соединения (сахароза, бензин, спирт).
Степень диссоциации. Константа диссоциации
Концентрация ионов в растворах зависит от того, насколько полно данный электролит диссоциирует на ионы. В растворах сильных электролитов, диссоциацию которых можно считать полной, концентрацию ионов легко определить по концентрации (c) и составу молекулы электролита (стехиометрическим индексам), например:
Концентрации ионов в растворах слабых электролитов качественно характеризуют степенью и константой диссоциации. Степень диссоциации (a) - отношение числа распавшихся на ионы молекул (n) к общему числу растворенных молекул (N):
a = n / N
и выражается в долях единицы или в % (a = 0,3 – условная граница деления на сильные и слабые электролиты).
Пример Определите мольную концентрацию катионов и анионов в 0,01 М растворах KBr, NH4OH, Ba(OH)2, H2SO4 и CH3COOH. Степень диссоциации слабых электролитов a = 0,3.
Решение KBr, Ba(OH)2 и H2SO4 - сильные электролиты, диссоциирующие полностью (a = 1).
KBr «K+ + Br- [K+] = [Br-] = 0,01 M
Ba(OH)2 «Ba2+ + 2OH- [Ba2+] = 0,01 M [OH-] = 0,02 M
H2SO4 «2H+ + SO4 [H+] = 0,02 M [SO42-] = 0,01 M
NH4OH и CH3COOH – слабые электролиты (a = 0,3)
NH4OH+4 + OH- [NH+4] = [OH-] = 0,3 • 0,01 = 0,003 M
CH3COOH «CH3COO- + H+ [H+] = [CH3COO-] = 0,3 • 0,01 = 0,003 M
Степень диссоциации зависит от концентрации раствора слабого электролита. При разбавлении водой степень диссоциации всегда увеличивается, т.к. увеличивается число молекул растворителя (H2O) на одну молекулу растворенного вещества. По принципу Ле Шателье равновесие электролитической диссоциации в этом случае должно сместиться в направлении образования продуктов, т.е. гидратированных ионов. Степень электролитической диссоциации зависит от температуры раствора. Обычно при увеличении температуры степень диссоциации растет, т.к. активируются связи в молекулах, они становятся более подвижными и легче ионизируются. Концентрацию ионов в растворе слабого электролита можно рассчитать, зная степень диссоциации a и исходную концентрацию вещества c в растворе.
Пример Определите концентрацию недиссоциированных молекул и ионов в 0,1 М раствора NH4OH, если степень диссоциации равна 0,01.
Решение Концентрации молекул NH4OH, которые к моменту равновесия распадутся на ионы, будет равна ac. Концентрация ионов NH4- и OH- - будет равна концентрации продиссоциированных молекул и равна ac (в соответствии с уравнением электролитической диссоциации)
[N+H4] = [OH]- = ac = 0,01 • 0,1 = 0,001 моль/л [NH4OH] = c - ac = 0,1 – 0,001 = 0,099 моль/л
Константа диссоциации (KD) - отношение произведения равновесных концентраций ионов в степени соответствующих стехиометрических коэффициентов к концентрации недиссоциированных молекул. Она является константой равновесия процесса электролитической диссоциации; характеризует способность вещества распадаться на ионы: чем выше KD, тем больше концентрация ионов в растворе. Диссоциации слабых многоосновных кислот или многокислотных оснований протекают по ступеням, соответственно для каждой ступени существует своя константа диссоциации:
Первая ступень: H3PO4 «H+ + H2PO4- KD1 = ([H+][H2PO4-]) / [H3PO4] = 7,1 • 10-3
Вторая ступень: H2PO4- «H+ + HPO42- KD2 = ([H+][HPO42-]) / [H2PO4-] = 6,2 • 10-8
Третья ступень: HPO42- «H+ + PO43- KD3 = ([H+][PO43-]) / [HPO42-] = 5,0 • 10-13
KD1 > KD2 > KD3
Пример Получите уравнение, связывающее степень электролитической диссоциации слабого электролита (a) с константой диссоциации (закон разбавления Оствальда) для слабой одноосновной кислоты НА.
HA «H+ + A+ KD = ([H+][A-]) / [HA]
Если общую концентрацию слабого электролита обозначить c, то равновесные концентрации Н+ и A- равны ac, а концентрация недиссоциированных молекул НА - (c - ac) = c (1 - a)
KD = (a • c • ac) / c(1 - a) = a2c / (1 - a)
В случае очень слабых электролитов (a £ 0,01)
KD = c • a2 или a = \é(KD / c)
Пример Вычислите степень диссоциации уксусной кислоты и концентрацию ионов H+ в 0,1 M растворе, если KD(CH3COOH) = 1,85 • 10-5
Решение Воспользуемся законом разбавления Оствальда
\é(KD / c) = \é((1,85 • 10-5) / 0,1)) = 0,0136 или a = 1,36% [H+] = a • c = 0,0136 • 0,1 моль/л
|
||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 655; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.14.145.167 (0.01 с.) |


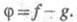 До тех пор пока g > f, будет наблюдаться цепная реакция, кинетика к-рой не отличается от неразветвленных цепных реакций. С ростом f в соответствии с (2) п будет нарастать до все более высоких стационарных значений:
До тех пор пока g > f, будет наблюдаться цепная реакция, кинетика к-рой не отличается от неразветвленных цепных реакций. С ростом f в соответствии с (2) п будет нарастать до все более высоких стационарных значений: станет положит. величиной, решением ур-ния (5) будет выражение:
станет положит. величиной, решением ур-ния (5) будет выражение:

 =0 - граничное для перехода от стационарного режима разветвленно-цепного процесса к нестационарному, протекающему с самоускорением (рис. 1). Чем быстрее р-ция, чем больше концентрация активных частиц, тем сильнее ускоряется р-ция. Т. обр., для разветвленных цепных реакций реализуется положит. oбратная связь.
=0 - граничное для перехода от стационарного режима разветвленно-цепного процесса к нестационарному, протекающему с самоускорением (рис. 1). Чем быстрее р-ция, чем больше концентрация активных частиц, тем сильнее ускоряется р-ция. Т. обр., для разветвленных цепных реакций реализуется положит. oбратная связь.
 =f - g); w - скорость цепной р-ции, t - время.
=f - g); w - скорость цепной р-ции, t - время. продолжение цепи определяется р-циями:
продолжение цепи определяется р-циями:

 , но и возникают еще два переносчика цепи
, но и возникают еще два переносчика цепи  и
и  Каждый из переносчиков цепи может либо погибнуть в р-циях обрыва, либо начать новое звено цепи, с образованием еще большего числа переносчиков цепи. Приведенная ниже схема развития цепного процесса в условиях, когда f > g, наглядно иллюстрирует явление разветвления цепи:
Каждый из переносчиков цепи может либо погибнуть в р-циях обрыва, либо начать новое звено цепи, с образованием еще большего числа переносчиков цепи. Приведенная ниже схема развития цепного процесса в условиях, когда f > g, наглядно иллюстрирует явление разветвления цепи:
 по р-ции
по р-ции  с послед. гибелью
с послед. гибелью  в р-циях друг с другом или с переносчиками цепи
в р-циях друг с другом или с переносчиками цепи  и
и  За счет тримолекулярных р-ций фактор g, пропорциональный р2, опережает в своем росте фактор f, пропорциональный р. В результате при нек-ром р2 - давлении верхнего предела -вновь происходит переход через граничное условие f = g и смесь теряет способность к самовоспламенению. Явление верхнего предела было открыто и объяснено С. Хиншелвудом (1956). Семенову и Хиншелвуду за исследование механизма хим. р-ций была присуждена Нобелевская премия.
За счет тримолекулярных р-ций фактор g, пропорциональный р2, опережает в своем росте фактор f, пропорциональный р. В результате при нек-ром р2 - давлении верхнего предела -вновь происходит переход через граничное условие f = g и смесь теряет способность к самовоспламенению. Явление верхнего предела было открыто и объяснено С. Хиншелвудом (1956). Семенову и Хиншелвуду за исследование механизма хим. р-ций была присуждена Нобелевская премия. 
 Другая возможность - переход энергии экзотермич. хим. р-ции в кинетич. энергию частиц и рост т-ры в случае, если скорость разогревания смеси превышает скорость отвода тепла от сосуда. Повышение т-ры приводит к ускорению р-ции, повышению интенсивности тепловыделения и дальнейшему росту т-ры и скорости р-ции - тепловому взрыву. В основе такого процесса м. б. как неразветвленная цепнаяреакция, так и р-ция с разветвлением цепей. В последнем случае появляется т. наз. третий предел самовоспламенения: смесь, потерявшая способность к самовоспламенению при р>р2, с послед. повышением р вновь становится самовоспламеняющейся.
Другая возможность - переход энергии экзотермич. хим. р-ции в кинетич. энергию частиц и рост т-ры в случае, если скорость разогревания смеси превышает скорость отвода тепла от сосуда. Повышение т-ры приводит к ускорению р-ции, повышению интенсивности тепловыделения и дальнейшему росту т-ры и скорости р-ции - тепловому взрыву. В основе такого процесса м. б. как неразветвленная цепнаяреакция, так и р-ция с разветвлением цепей. В последнем случае появляется т. наз. третий предел самовоспламенения: смесь, потерявшая способность к самовоспламенению при р>р2, с послед. повышением р вновь становится самовоспламеняющейся.  2HF;
2HF;  = -537 кДж/моль
= -537 кДж/моль 2НС1 протекает как неразветвленный процесс. В отсутствие светового воздействия для обеих р-цийинициирование происходит на стенке сосуда: (Х2 + S
2НС1 протекает как неразветвленный процесс. В отсутствие светового воздействия для обеих р-цийинициирование происходит на стенке сосуда: (Х2 + S  X2S
X2S  SX +
SX +  ) и в объеме (Х2 + М
) и в объеме (Х2 + М  М + 2Х), где X - С1 или F. Большая разница энергий связи F — F (159 кДж/моль) и Н — F (565 кДж/моль) определяет еще один канал образования атомарного F:
М + 2Х), где X - С1 или F. Большая разница энергий связи F — F (159 кДж/моль) и Н — F (565 кДж/моль) определяет еще один канал образования атомарного F:


 (значок v означает колебат. возбуждение). Частица
(значок v означает колебат. возбуждение). Частица  как и Н2, реагирует с F2, но для р-ции
как и Н2, реагирует с F2, но для р-ции  тепловой эффект составляет не +30, а -22,7 кДж/моль, а ее константа скорости примерно в 107 больше, чем для р-ции невозбужденного Н2. Дополнив р-ции 7 и 2 рассмотренными р-циями колебательно-возбужденных частиц, получаем след. ЦПЦ:
тепловой эффект составляет не +30, а -22,7 кДж/моль, а ее константа скорости примерно в 107 больше, чем для р-ции невозбужденного Н2. Дополнив р-ции 7 и 2 рассмотренными р-циями колебательно-возбужденных частиц, получаем след. ЦПЦ:

 CF4 + 2HF был установлен механизм разветвления цепей в результате распада промежут. в-ва, образующегося в состоянии колебат. возбуждения:
CF4 + 2HF был установлен механизм разветвления цепей в результате распада промежут. в-ва, образующегося в состоянии колебат. возбуждения:

 =f - g таковы, что вся р-ция протекает за доли секунды.
=f - g таковы, что вся р-ция протекает за доли секунды. Для таких р-ций обычно не наблюдается перехода в режим самовоспламененияили взрыва. По достижении нек-рой макс. скорости такие р-ции замедляются (рис. 3). С этим связано второе назв. таких р-ций - вырожденный цепной взрыв. Характерное время их ускоренного протекания - не доли секунды, а десятки минут и часов. Различие величин j определяет различие механизмов.
Для таких р-ций обычно не наблюдается перехода в режим самовоспламененияили взрыва. По достижении нек-рой макс. скорости такие р-ции замедляются (рис. 3). С этим связано второе назв. таких р-ций - вырожденный цепной взрыв. Характерное время их ускоренного протекания - не доли секунды, а десятки минут и часов. Различие величин j определяет различие механизмов.


 не участвуют в ЦПЦ и исчезают в разл. р-циях гибели на стенке и в объеме. Концентрации частиц
не участвуют в ЦПЦ и исчезают в разл. р-циях гибели на стенке и в объеме. Концентрации частиц  и
и  за доли секунды достигают своего стационарного значения, определяемого ур-нием (2), a ROOH накапливается в системе и служит инициатором:
за доли секунды достигают своего стационарного значения, определяемого ур-нием (2), a ROOH накапливается в системе и служит инициатором:
 происходит в результате р-ций:
происходит в результате р-ций: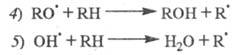


 и
и  происходил с характерным временем, соизмеримым со временем развития звена цепи, не было бы отличия в их поведении. В действительности же эти времена сильно разнятся. Одна р-ция разветвления 3 приходится на многие тысячи р-ций 1 и 2, составляющих ЦПЦ неразветвленного окисления RH. Рост [ROOH] в ходе р-ции мог бы тем не менее привести к тому, что величины k3[ROOH] и
происходил с характерным временем, соизмеримым со временем развития звена цепи, не было бы отличия в их поведении. В действительности же эти времена сильно разнятся. Одна р-ция разветвления 3 приходится на многие тысячи р-ций 1 и 2, составляющих ЦПЦ неразветвленного окисления RH. Рост [ROOH] в ходе р-ции мог бы тем не менее привести к тому, что величины k3[ROOH] и  стали бы соизмеримы. Этого не происходит из-за того, что ROOH - нестабильный промежут. продукт, и со скоростью, намного большей, чем для р-ции 5, происходит превращение ROOH в стабильные продукты окисления RH - кетоны, к-ты и др. по р-ции:
стали бы соизмеримы. Этого не происходит из-за того, что ROOH - нестабильный промежут. продукт, и со скоростью, намного большей, чем для р-ции 5, происходит превращение ROOH в стабильные продукты окисления RH - кетоны, к-ты и др. по р-ции:
 не происходит значит, накапливания ROOH, а по мере его накапливания расход RH по р-циям 1, 2 и 6 оказывается столь большим, что скорость всех р-ций с участием RH падает значительно. Рассмотренными факторами и определяются характерные особенности вырожденно-разветвленных цепных реакций: рост скорости на начальных стадиях, описываемый ур-нием
не происходит значит, накапливания ROOH, а по мере его накапливания расход RH по р-циям 1, 2 и 6 оказывается столь большим, что скорость всех р-ций с участием RH падает значительно. Рассмотренными факторами и определяются характерные особенности вырожденно-разветвленных цепных реакций: рост скорости на начальных стадиях, описываемый ур-нием  но с очень малым
но с очень малым  уменьшение скорости р-ции на более поздних стадиях р-ции. К р-циям с вырожденным разветвлением относятся многочисленные р-ции окисленияне только углеводородов, но и большинства др. орг. соединений, поскольку их общим св-вом является промежут. образование ROOH. Подобным образом протекает и окисление сероводорода.
уменьшение скорости р-ции на более поздних стадиях р-ции. К р-циям с вырожденным разветвлением относятся многочисленные р-ции окисленияне только углеводородов, но и большинства др. орг. соединений, поскольку их общим св-вом является промежут. образование ROOH. Подобным образом протекает и окисление сероводорода.  на
на  и альдегид R"CHO и последующей р-ции
и альдегид R"CHO и последующей р-ции 




