
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Основные типы гетерогенных катализаторовСодержание книги
Поиск на нашем сайте
В Г. к., как и в др. областях катализа, выделяют два типа р-ций: окислительно-восстановительные, при к-рых роль катализатора сводится к участию в переносе неспаренных электронов, и кислотно-основные, при к-рых взаимод. катализатора с реагирующими в-вами сопровождается переходом протона или электронных пар. Окислит.-восстановит. Г. к. происходит на пов-сти металлов или полупроводников, т.е. в-в, способных передавать или принимать неспаренные электроны от реагирующих молекул. Кислотно-основные р-ции протекают на пов-сти твердых к-т или оснований, способных передавать или принимать протон от реагентов или же способных к хим. взаимод. с реагентами без разделения пары электронов. Рассмотрим возможные механизмы этих взаимодействий. Катализаторы-полупроводники. Согласно электронной теории Г. к., каталитич. активность полупроводников связана с объемной концентрацией носителей тока (электронов и дырок). Адсорбция частицы на пов-сти полупроводника приводит к образованию дополнит. (примесного) энергетич. уровня в запрещенной зоне. Переход электрона или дырки на этот уровень изменяет их объемную концентрацию и св-ва пов-сти (напр., работу выхода электрона), на к-рой возникают заряженные центры, участвующие в каталитич. превращении. Можно представить, напр., что дегидрирование изопропилового спирта происходит по механизму: где е-электрон катализатора, где а и b- эмпирии, постоянные. Наиб. важную роль в Г. к. играют полупроводники, представляющие собой соед. элементов VI гр. периодич. системы (О, S, Se, Те) с переходными металлами (обычно оксиды или сульфиды металлов). Каталитич. св-ва этих в-в определяются наличием у атомов переходных металлов неск. степеней окисления, к-рые в условиях катализа легко изменяются в результате переноса электрона от атома др. реагента. Напр., Мо в оксидах может иметь степени окисления Мо2 +, Мо3 +, Мо4 +, Мо5+ и Мо6+, поэтому он легко окисляется и восстанавливается в условиях Г. к., обеспечивая тем самым каталитич. цикл. Важно также наличие у поверхностных атомов переходных металлов низкоэнергетич. d-орбиталей разной симметрии. Это позволяет связать каталитич. активность их оксидов и сульфидов с электронной конфигурацией J-орбиталей, а также с возможностью образования промежут. поверхностных соед. типа комплексных. Напр., для оксидов металлов установлена четкая корреляция между константами скорости H-D обмена и мн. р-ций окисления с числом d-электронов катиона, т. е. положением переходного элемента в периодич. системе. Максимальна активность оксидов с электронной конфигурацией катионов d3 (Cr2O3, МnО,) и d6-d8 (Co3O4, NiO), минимальна - с конфигурацией do (TiO2), d5 (Fe2O3), d10 (ZnO, Cu2O) (рис. З). Подобная зависимость объясняется кристаллического поля теорией.
Рис. 3. Изменение каталитич. активности оксидов переходных металлов в р-ции обмена H2-D2. Методами ИК-спектроскопии подтверждено, что в ряде р-ций с участием олефинов (селективное окисление, полимеризация, диспропорционирование) на пов-сти оксидов переходных металлов с электронной конфигурацией катиона d1 (Мо5 +, V4 +, Ti3 +) образуются Катализаторы-металлы. Металлы обычно значительно активнее оксидов и обладают более универсальным каталитич. действием, хотя, как правило, менее селективны. наиб. универсальны металлы VIII гр. периодич. системы, особенно Pt и Pd, катализирующие разл. р-ции окисления, гидрирования, дегидрирования и т.д. при низких т-рах (комнатной и более низких). Каталитич. активность определяется электронной конфигурацией и симметрией d- орбиталей поверхностных атомов. В хим. взаимод. с молекулами реагирующих на пов-сти в-в участвуют только те d- орбитали, к-рые направлены от пов-сти наружу и имеют одинаковую группу симметрии с молекулярными орбиталями реагентов. Участие d-электронов в хим. связи металла с адсорбиров. молекулами подтверждено методами фотоэлектронной и УФ-спектроскопии для Pt-катализатора.
Металлы, находящиеся в конце переходных периодов, имеют в d-оболочке дырки (отсутствие электронов), что облегчает их участие в каталитич. превращении. Металлы, находящиеся в начале периода, обычно образуют прочную связь с молекулами реагентов. Это приводит к образованию фаз (поверхностных или объемных) оксидов, гидридов и т. п. и снижению каталитич. активности металла. Так, Ni активен в р-циях гидрирования, а Си малоактивна. При сплавлении активных металлов VIII группы с неактивными металлами 1б группы каталитич. активность уменьшается вследствие заполнения d-оболочки электронами. Напр., для сплавов Cu-Ni падение активности наступает при составе 53% Си и 47% Ni, когда s-электроны Си заполняют J-оболочку Ni. В р-циях с участием Н2 наиб. активны металлы, на пов-сти к-рых происходит его хемосорбция с диссоциацией и низкой энергией связи атомарного водорода. Сплавы Cu-Ni, Au-Pt, Ag-Pd менее активны, чем чистые металлы VIII группы. На чистых металлах 1б группы Н2 не адсорбируется и не активируется. В пром-сти широко применяют мелкодисперсные метал-лич. катализаторы, нанесенные на носители (SiO2, A12O3, алюмосиликаты, активный уголь, кизельгур и др.). Это повышает пов-сть катализатора, уменьшает его расход, предохраняет частицы от спекания. По мере уменьшения размера частиц (повышения дисперсности) катализатора его активность в одних р-циях остается неизменной (структурно-нечувствительные или "незатрудненные" р-ции), в других р-циях на частицах размером 2-4 нм при общем росте активности наблюдается снижение числа оборотов р-ции; это т. наз. структурно-чувствительные или "затрудненные" р-ции. Так, большинство р-ций гидрирования олефинов и ароматич. соед. являются структурно-нечувствительными; р-ции синтеза NH3, гидрогенолиза связи С—С, окисления, изомеризации и др. структурно-чувствительны. Различие между этими типами гетерогенно-каталитич. р-ций на металлах объясняют тем, что в первом из них активным центром является каждый атом пов-сти, во втором-совокупность неск. атомов. Разбавление в катализаторе-сплаве активного металла неактивным компонентом не влияет на его активность в структурно-нечувствит. р-циях и приводит к значит, ее снижению в структурно-чувствит. р-циях. Кроме того, в структурно-чувствительных р-циях активность катализатора зависит от выхода на пов-сть кристаллич. граней, ребер, концентрации дислокаций, Так, в синтезе NH3 на монокристаллах Fe наиб. активностью обладает грань (111), в гидрогенолизе углеводородов на Pt и Pd-ребра монокристаллов; необходимый для р-ции Н2диссоциирует на ребрах и диффундирует к активным центрам, на к-рых происходит каталитич. превращение. Каталитич. активность металлов на носителях изменяется также вследствие их хим. взаимод. с носителем (см. Нанесенные катализаторы). Катализ на твердых кислотах и основаниях. Для катализаторов кислотно-основного типа специфика твердого тела не выражена так резко, как для полупроводников и металлов. Активные центры кислотных кат. представляют подвижные протоны Н (центры Бренстеда) или атомы, способные присоединять пару электронов (центры Льюиса), напр. атом А1 на пов-сти А12О3. Соотв. основными центрами являются акцепторы протона или доноры электронной пары, напр. атомы кислорода на пов-сти CaO, MgO и т.п. Кислотными бренстедовскими центрами простых оксидов металлов являются поверхностные гидроксильные группы, остающиеся после частичной дегидратации пов-сти при нагр., или молекулы Н2О, координационно связанные с пов-стью. Для металла М, находящегося в начале каждого периода, гидроксильные группы имеют основные св-ва [...ОМ]+ [ОН]-, для находящегося в конце периода-кислотные: [...ОМО]-Н +. Льюисовскими кислотными центрами служат координационно-ненасыщенные ионы, напр. АlO-2 на А12О3. Эти центры способны взаимод. с реагирующей молекулой-донором пары электронов. Кислотными катализаторами являются оксиды металлов с большим отношением заряда иона к его радиусу-оксиды Мо, Zn, Ca, Pb и др. Их активность связана с положением металла в периодич. системе и возрастает в периодах при переходе к V-VII группам, а в группах-при переходе к I периоду.
Смешанные катализаторы. В р-циях кислотно-основного типа (крекинг, дегидратация, изомеризация и др.) высокой активностью обладают катализаторы, состоящие из неск. в-в,- оксиды металлов с разл. зарядом катиона, аморфные алюмосиликаты и цеолиты, гетерополикислоты, сульфаты, фосфаты и др. Именно на пов-сти смешанных систем легче образуются реакционноспособные заряженные частицы. Напр., в алюмосиликатах ион А13+ замещает Si4+ в кремнекислородной решетке; меньший заряд Al3+ по сравнению с Si4+ компенсируется появлением центра Бренстеда Н+. Присоединение образовавшегося Н + к реагентам приводит к возникновению заряженных реакционноспособных частиц, напр. карбкатионов (СН3СН=СН2 + Н+ -> C3H7+), участвующих далее в катализе. На основных центрах образуются отрицательно заряженные частицы, в р-циях углеводородов-карбанионы. Полифункциональные катализаторы. Пром. каталитич. процессы часто проводят на катализаторах, сочетающих разл. ф-ции. Напр., превращения углеводородов в риформинге ускоряются катализаторами, в к-рых переходные металлы, гл.обр. Pt или Ni, комбинируются с кислотным оксидом, напр. алюмосиликатом или А12О3, модифицированным фтором. В этом случае Pt оказывает дегидрирующее действие, а кислотный оксид-изомеризующее. Катализ протекает вблизи границы раздела фаз или в результате перемещения активной частицы из одной фазы в другую: Полифункциональные кат., как правило, состоят из неск. фаз, каждая из к-рых ускоряет одну из стадий сложного процесса. Более 50 лет назад С. В. Лебедев совм. с сотрудниками подобрал селективный кат. синтеза бутадиена из спирта по р-ции: 2С2Н5ОН -> СН2=СНСН=СН2 + 2Н2О + Н2 Были выделены стадии процесса: дегидратация, дегидрирование и конденсация и для каждой из стадий подобран свой катализатор (активная глина, ZnO и MgO соотв.). В смешанном кат. эти компоненты находятся в соотношении, обеспечивающем макс. выход бутадиена.
Высокоселективные кат. парциального окисления представляют смесь оксидов разл. металлов. Напр., для окисления пропилена в акролеин применяют катализатор, состоящий из оксидов Bi, Mo, Fe, Co и др. В этом катализаторе Bi2(MoO4) служит для адсорбции и активации пропилена, FеМоО4-для активации О2, на дефектах кристаллич. структуры Fe2(MoO4)3 происходит перенос ионов кислорода от центров его адсорбции к центрам адсорбции олефина, СоМоО4 служит для стабилизации структуры FeMoO4. Гетерогенизированньзе металлокомплексные катализаторы. В 70-80-е гг. 20 в. широко исследуются катализаторы-комплексы металлов, закрепленные на пов-сти носителя (SiO2, А12О3 и др.). Состав таких комплексов описывается общей ф-лой XnMmYy, где М-активный центр (атом) переходного металла, Х-лиганд, связывающий атом металла с пов-стью, Y-внеш. лиганд. В общем случае комплекс м. б. моноядерным (т = 1) или полиядерным (т При взаимод. металлоорг. соед. с ОН-группами носителя можно получить закрепленные комплексы, не имеющие р-римых аналогов. Гетерогенизированные металлокомплексные кат. сочетают высокую активность и однородность по каталитич. св-вам, характерные для гомог. катализа, с удобствами технол. применения гетерог. катализаторов. Получены Гетерогенизированные кат., содержащие комплексы Ti, Zr, Mo, W, Ni и др. металлов, активные в полимеризации, окислении, гидрировании и др. Технол. применение гетерогенизированных комплексных кат. пока невелико из-за нестабильности гетерогенизиров. комплексов в условиях катализа и трудности их регенерации. Гетерогенизиров. комплексы используют для получения нанесенных металлич. катализаторов очень высокой дисперсности (размер частички - кластера - неск. атомов).
|
||||||||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 326; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.118.255.51 (0.013 с.) |

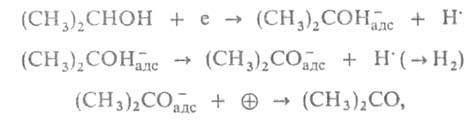
 -своб. дырка. Поскольку объемная концентрация носителей тока зависит от положения уровня Ферми и изменяется при всяком сдвиге последнего, предполагалась возможность регулирования каталитич. активности полупроводника смещением уровня Ферми. Дальнейшие исследования, однако, не подтвердили суще" ствования электронного равновесия между пов-стью и объемом катализатора-полупроводника в условиях Г. к. Экспериментально установлен ряд корреляций каталитич. активности полупроводников с проводимостью п- или р -типа с их св-вами. В частности, известна корреляция константы скорости Г. к. с шириной запрещенной зоны
-своб. дырка. Поскольку объемная концентрация носителей тока зависит от положения уровня Ферми и изменяется при всяком сдвиге последнего, предполагалась возможность регулирования каталитич. активности полупроводника смещением уровня Ферми. Дальнейшие исследования, однако, не подтвердили суще" ствования электронного равновесия между пов-стью и объемом катализатора-полупроводника в условиях Г. к. Экспериментально установлен ряд корреляций каталитич. активности полупроводников с проводимостью п- или р -типа с их св-вами. В частности, известна корреляция константы скорости Г. к. с шириной запрещенной зоны  :
: 

 комплексы и
комплексы и  аллильные комплексы типа I, где М-атом металла.
аллильные комплексы типа I, где М-атом металла.
 2) и связан с пов-стью одним или неск. лигандами X. Напр., растворимый комплексный катализатор гидрирования Rh[P(C6H5)3]3Cl м. б. закреплен на пов-сти силикагеля:
2) и связан с пов-стью одним или неск. лигандами X. Напр., растворимый комплексный катализатор гидрирования Rh[P(C6H5)3]3Cl м. б. закреплен на пов-сти силикагеля: 



