
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
U – изохорно-изэнтропный термодинамический потенциал.Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
H = U + PV dH = dU + PdV + VdP, dU £ TdS – PdV dH £ TdS + VdP, H = H (S,P) dH = Т – мера приращения Н с ростом S при P = const V – мера приращения H с ростом P при S = const. При S,P = const: (¶H)S,P £ 0 (¶H)S,P < 0 – критерий направления процесса в условиях S,P = const; (¶H)S,P = 0, (¶ 2H)S,P > 0 – критерий достижения равновесия. Н – изобарно-изэнтропный потенциал. U, Н – эти функции могут служить критерием равновесия при S = const. S непосредственно измерить нельзя, и контроль ее постоянства при неравновесных процессах затруднителен. Поэтому функции U и Н не находят широкого применения в качестве критериев направления процесса и равновесия. Сопоставим выражения для полных дифференциалов функций U, H, F и G:
Частными производными этих четырех функций при данном, характерном для каждой из них наборе независимых переменных являются основные параметры состояния системы: Р, V, T, S. Отсюда вытекает важное свойство этих функций: через каждую из этих функций и ее производные можно выразить в явной форме любое термодинамическое свойство системы. Каждая функция дает, таким образом, полную термодинамическую характеристику системы. Поэтому эти функции (термодинамические потенциалы) называют характеристическими функциями. Каждая функция обладает свойствами характеристической функции только при соответствующем, характерном для нее наборе независимых переменных. Поэтому независимые переменные в вышеприведенных уравнениях называют естественными переменными. Энтропию также можно отнести к этим функциям (может служить критерием направления процесса и достижения равновесия): dQ = dU + dA £ TdS, TdS ³ dU + PdV, S = S (U,V) (¶S)U,V > 0 – критерий самопроизвольного протекания процесса в условиях U,V = const; (¶S)U,V = 0, (¶ 2S)U,V < 0 – критерий равновесия. Т.о., условия равновесия системы можно сформулировать следующим образом: в состоянии равновесия системы ее термодинамические потенциалы имеют минимальное значение при постоянстве своих естественных переменных, а энтропия имеет максимальное значение. Другое общее свойство термодинамических потенциалов – то, что убыль их в равновесном процессе при постоянстве естественных переменных равна максимальной полезной работе. Лекция 7 Уравнения Гиббса-Гельмгольца. Статистический характер второго закона термодинамики.
Уравнения максимальной работы (уравнения Гиббса-Гельмгольца). Свойства функций F и G дают возможность установить связь между максимальной работой процесса, протекающего равновесно, и теплотой того же процесса, но протекающего неравновесно.
Запишем это уравнение для двух состояний системы: F1 = U1 + T F1 – F2 = U1 – U2 + T Amax = Видно: зная Amax (или изменение изохорного потенциала) и зависимость ее от Т, можно вычислить теплоту процесса (т.е. изменение внутренней энергии). Если же известна Q процесса, то для расчета Amax необходимо интегрировать уравнение (1), причем появляется постоянная интегрирования, для определения которой необходимы дополнительные сведения. Уравнение (1) – уравнение максимальной работы (уравнение Гиббса-Гельмгольца) – может быть записано и в форме: DF = DU + T Другие варианты уравнения Гиббса-Гельмгольца:
G1 = H1 + T G1 – G2 = H1 – H2 + T A¢max = DG = DH + T DU = QV и DH = QP справедливы, если в 1-м случае не совершается никакая работа (А = 0), а во 2-м случае совершается только работа расширения (А¢ = 0). Поэтому в уравнениях (1) и (3) теплоты Исходя из вышеизложенного, эти уравнения можно записать так: – DF = Amax = – QV, неравн + QV, равн – DG = A¢max = – QP, неравн + QP, равн
СТАТИСТИЧЕСКИЙ ХАРАКТЕР II ЗАКОНА ТЕРМОДИНАМИКИ. Термодинамический метод неприменим к системам, состоящим из малого числа молекул (единицы, десятки, сотни). Это связано с тем, что в таких системах исчезает различие между теплотой и работой. Одновременно исчезает однозначность направления процесса, устанавливаемая вторым законом термодинамики. Категорическое утверждение о невозможности одного из направлений процесса заменяется оценкой относительной вероятности обоих противоположных направлений. Для очень малого числа молекул оба направления процесса становятся равноценными. Чисто механическое движение отдельных молекул обратимо и преимущественного направления не имеет. В отличие от I закона термодинамики, который является абсолютным, II закон термодинамики имеет статистический характер, он справедлив лишь для систем, имеющих большое число частиц. Ограничения перевода теплоты в работу, которые устанавливаются вторым законом термодинамики, в противоположность неограниченному переводу работы в теплоту, связаны со стремлением системы, содержащей большое число частиц, к молекулярному беспорядку, мерой которого служит энтропия. Работа характеризуется упорядоченным коллективным движением частиц в одном направлении; теплота связана с хаотическим молекулярным движением. Поэтому выясняется самопроизвольный характер перехода работы в теплоту, как перехода молекулярной системы к более вероятному состоянию, которому отвечает большая степень молекулярного беспорядка. Причиной стремления к равновесию является переход молекулярной системы от менее вероятного состояния к более вероятному состоянию большего молекулярного беспорядка; при этом возрастает энтропия. Связь между S и вероятностью состояния системы можно установить таким образом: любое термодинамическое макросостояние системы может быть осуществлено различными микросостояниями. Число микросостояний, с помощью которых можно осуществить данное макросостояние системы, называется термодинамической вероятностью W. Число микросостояний можно рассчитать по формуле: W = N – общее число молекул, распределенных либо по k отделениям, либо по k уровням, либо по скоростям и т.д.; N1 – число молекул в 1-м отделении; N2 – число молекул во 2-м отделении … Рассмотрим, как меняется W по мере протекания процесса. Самопроизвольный процесс сопровождается возрастанием S; при этом резко возрастает также W. Следовательно, S и W должны быть связаны друг с другом: S = S (W) Надо учесть аддитивность S: S = S1 + S2 Для W: W = W1 × W2 (вероятность события равна произведению вероятностей отдельных независимых событий). Если равновесная система с вероятностью W и энтропией S состоит из двух частей с вероятностями W1 и W2 и энтропиями S1 и S2, то можно записать: S1 = f (W1), S2 = f (W2) S = f (W) = f (W1×W2), S1 + S2 = f (W1) + f (W2) f (W) = f (W1×W2) = f (W1) + f (W2) Для системы из многих частей: f (W) = f (W1×W2× … W k) = f (W1) + f (W2) + … + f (W k) Решением этого уравнения является уравнение Больцмана: S = k ln W k – постоянная Больцмана; k = R / NA Рассмотрим процесс изотермического расширения 1 кмоля идеального газа.
Для N молекул: W¢N =
S2 – S1 = k ln W¢¢N – k ln W¢N, S = k ln W + const сonst = 0 – постулат Больцмана Þ S = k ln W Это соотношение вскрывает статистический характер II закона термодинамики. Условие возрастания S в изолированной системе не обязательно, а вероятно. Возможны самопроизвольные отрицательные процессы, сопровождающиеся уменьшением S в изолированной системе, например, флуктуации плотности (не только возможны, но и повсеместно осуществляются). Значительные отклонения от средних величин в больших системах имеют исчезающе малую вероятность, но в принципе они также возможны. Установление статистического характера II закона термодинамики является великой заслугой Больцмана, объяснившего таким образом противоречие между обратимостью механического движения и необратимостью и направленностью реальных физических и химических процессов. Эта направленность является следствием молекулярного строения материального мира. В работах Больцмана, Смолуховского и др. ученых показан статистический характер II закона термодинамики и количественно изучены наблюдаемые отклонения от этого закона. Этими работами окончательно показана несостоятельность антинаучной идеи тепловой смерти вселенной, высказанной Клаузиусом. Клаузиус неправильно толковал II закон термодинамики (одним из творцов которого он был), как абсолютный закон природы. Незаконно распространяя свой постулат на вселенную, которую он уподоблял изолированной системе, и на неограниченный промежуток времени, Клаузиус дал второму закону термодинамики следующую формулировку: энтропия вселенной стремится к максимуму. Из этого положения вытекают два вывода: 1. Через достаточно большой промежуток времени вселенная приблизится к такому состоянию, что ее энтропия будет близка к максимальной величине; состояние вселенной будет близко к равновесному и все процессы угаснут – останутся равномерно распределенные в пространстве материя и энергия. Дальнейшая эволюция вселенной прекратится, наступит «тепловая смерть вселенной». 2. Т.к. в настоящее время вселенная далека от «тепловой смерти», хотя и движется только в направлении к ней, то, следовательно, вселенная имела начало, она возникла в противоречии со II законом термодинамики (имеющим абсолютное значение) в результате какого-то творческого акта, не подчиняющегося законам природы. Выводы Клаузиуса о тепловой смерти вселенной незакономерны, т.к. термодинамические свойства конечной изолированной системы распространялись им на вселенную, безграничную в пространстве и времени. Работы Больцмана и др. ученых, установивших ограниченный статистический характер II закона термодинамики, показали возможность и необходимость наличия во вселенной любых по величине отклонений от требований второго закона для равновесных систем. Само представление о движении вселенной (как целого) к равновесию незакономерно.
Лекция 8 Термодинамические потенциалы идеальных и реальных газов. Летучесть и методы ее расчета.
1. Внутренняя энергия 1 моля идеального газа, зависящая только от Т, выражается уравнением: dU = CV dT, U = Uo + 2. Энтальпия: H = U + PV = Uo + 3. Энтропия: S = CV ln T + R ln V + So¢, S = CP ln T - R ln P + So 4. dF = – PdV – SdT, dG = VdP – SdT Пусть T = const: dF = – PdV = – F = – RT ln V + F(T), G = RT ln P + G(T) (т.к. интегрирование проведено при T = const, то постоянные интегрирования – функции Т).
фугитивность. f подставляется в термодинамические уравнения вместо Р. G = RT ln f + G(T) (1) При этом вводится условие: Т.о., метод Льюиса – это математический прием, который состоит в введении новой функции f, промежуточной между параметрами состояния газа Р и Т, с одной стороны, и G – с другой стороны. DG = G2 – G1 = RT ln f 2 / f 1 при T = const Чтобы вычислить DG для процессов с реальным газом, нужно знать зависимость f от Р и Т. Уравнение (1) и условие (2) – это основа для вычисления f. Продифференцируем уравнение (1) по Р при Т = const:
Учитывая, что
Надо знать зависимость V от Р. Наиболее точный способ заключается в графическом нахождении интеграла этого уравнения. Для этого по экспериментальным значениям V, который занимает 1 моль газа при разных Р, строят кривую зависимости V от Р. Интеграл равен площади под соответствующей частью кривой. Имея опытные данные V = V(P), вычисляют так называемую объемную поправку реального газа a, определяемую по уравнению: V = ln ln f 2 – ln f 1 = ln P2 – ln P1 – Пусть нижний предел интегрирования Р1 очень мал, так чтобы f 1 = P1: ln f = ln P – ln g =
Если Т – высокие, Р – невысокие, a = const, тогда: ln f = ln P – При малых a и Р функция может быть разложена в ряд Маклорена; ограничимся двумя членами ряда:
(
Вышеприведенное уравнение дает возможность приближенно вычислять f при малых Р. Кроме того, для приближенного вычисления летучестей реальных газов можно воспользоваться методом расчета, основанном на принципе соответственных состояний. Согласно этому принципу, ряд одинаковых свойств, в том числе и коэффициент активности различных реальных газов, оказываются равными при одинаковых приведенных температурах (t) и приведенных давлениях (p): t = Т/Ткр, p = Р/Ркр
Лекция 9 Фазовые переходы. Уравнение Клапейрона-Клаузиуса. Фазовые переходы 1-го и 2-го рода. Зависимость давления насыщенного пара от температуры. Закон смещения равновесия.
В системе, состоящей из нескольких фаз чистого вещества, находящихся в равновесии, возможны переходы вещества из одной фазы в другую – это фазовые переходы (или превращения агрегатных состояний). Рассмотрим равновесный переход 1 моля вещества из фазы 1 в фазу 2 при Р,Т = const (жидкость – газ, например). U2 – U1 = Q – A = T (S2 – S1) – P (V2 – V1) (производится только работа расширения) U2 – TS2 + PV2 = U1 – TS1 + PV1 G2 = G1 Изобарные потенциалы единицы массы чистого вещества в двух фазах, находящихся в равновесии, равны между собой. Напишем для полных дифференциалов: dG1 = V1dP – S1dT, dG2 = V2dP – S2dT dG2 – dG1 = (V2 – V1)dP – (S2 – S1)dT = 0 (= 0, т.к. сохраняется фазовое равновесие и dG1 = dG2)
Т.к. процесс равновесный и изотермический, то можно записать: S2 – S1 = Q – теплота, поглощаемая при переходе 1 моля вещества из одной фазы в другую; DН – теплота фазового превращения; Т – температура фазового превращения; V2 – V1 – разность мольных объемов двух фаз.
Вышеприведенное уравнение (две формы записи) – уравнение Клапейрона-Клаузиуса – общее термодинамическое уравнение, приложимое ко всем превращениям агрегатных состояний чистых веществ. ФАЗОВЫЕ ПЕРЕХОДЫ ПЕРВОГО РОДА. Фазовые переходы, характеризующиеся равенством изобарных потенциалов двух сосуществующих в равновесии фаз и скачкообразным изменением S и V при переходе вещества из одной фазы в другую – фазовые переходы первого рода. К ним относятся плавление, испарение, возгонка, переход из одной кристаллической модификации в другую. ПЛАВЛЕНИЕ. DНпл всегда > 0; Vж > Vтв для большинства веществ (кроме воды, висмута и немногих др.), т.е. плотность твердой фазы больше плотности жидкой. Следовательно, ИСПАРЕНИЕ (ВОЗГОНКА). (DНисп всегда > 0; Vг > Vж для всех веществ, и поэтому Ткип всегда повышается с ростом Т). При Т, далекой от критической, Vг >> Vж, и значением Vж в уравнении можно пренебречь. DНисп = Т
|
||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-23; просмотров: 508; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.128.201.71 (0.009 с.) |

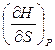 dS +
dS + 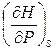 dP;
dP;  = – S, F = U – TS, F = U + T
= – S, F = U – TS, F = U + T  , F2 = U2 + T
, F2 = U2 + T 
 (F1 – F2)V = – DF
(F1 – F2)V = – DF + T
+ T 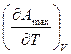 (1)
(1) (2)
(2) = – S, G = H – TS, G = H + T
= – S, G = H – TS, G = H + T 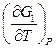 , G2 = H2 + T
, G2 = H2 + T 
 + T
+ T 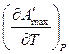 (3)
(3) (4)
(4)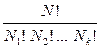

 =
=  = R ln
= R ln 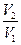 = k N ln
= k N ln  – вероятность нахождения одной молекулы в объеме V1
W¢¢1 =
– вероятность нахождения одной молекулы в объеме V1
W¢¢1 =  – вероятность нахождения одной молекулы в объеме V2
– вероятность нахождения одной молекулы в объеме V2
 , W¢¢N =
, W¢¢N = 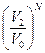
 =
= 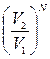 , ln
, ln 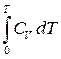
 = Uo +
= Uo + 
 dV, dG = VdP =
dV, dG = VdP =  dP
dP
 lim
lim  = 1 при Р ® 0 (2)
= 1 при Р ® 0 (2)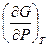 = RT
= RT 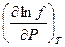
 =
=  , ln
, ln  =
= 

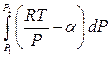 = ln
= ln  –
–  , надо знать a = a (Р).
, надо знать a = a (Р).

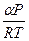 , g =
, g =  (
( =
= 


 =
= 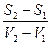
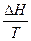
 , DН = Т
, DН = Т 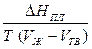
 =
=  > 0 и с ростом Р температура плавления повышается.
> 0 и с ростом Р температура плавления повышается. > 0
> 0


