
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Термохимия.Тепловой эффект реакции. Закон Гесса.Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Внутренняя энергия Определяется в соответствии с первым началом термодинамики, как разность между количеством теплоты, сообщенным системе, и работой, совершенной системой над внешними телами:
Энтальпия Определяется следующим образом:
где Поскольку в изобарном процессе работа равна Энтропия (S) – термодинамическая функция состояния, которая служит мерой беспорядка (неупорядоченности) системы. Возможность протекания эндотермических процессов обусловлена изменением энтропии, ибо в изолированных системах энтропия самопроизвольно протекающего процесса увеличивается Δ S > 0 (второй закон термодинамики). Л. Больцман определил энтропию как термодинамическую вероятность состояния (беспорядок) системы W. Поскольку число частиц в системе велико (число Авогадро N A = 6,02∙10 23), то энтропия пропорциональна натуральному логарифму термодинамической вероятности состояния системы W:
Размерность энтропии 1 моля вещества совпадает с размерностью газовой постоянной R и равна Дж∙моль –1∙K –1. Изменение энтропии *) в необратимых и обратимых процессах передается соотношениями Δ S > Q / T и Δ S = Q / T. Например, изменение энтропии плавления равно теплоте (энтальпии) плавления Δ S пл = Δ H пл/ T пл Для химической реакции изменение энтропии аналогично изменению энтальпии
Энергия Гиббса Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
где Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.) 2. Термохимия.Тепловой эффект реакции. Закон Гесса. Закон Гесса может быть сформулирован так: тепловой эффект реакции, состоящей из нескольких промежуточных стадий,не зависит от этих промежуточных стадий или их последовательности, а полностью определяется начальным и конечным состояниями системы.
Закон Гесса может быть выражен также следующим образом: если система посредством ряда химических превращений совершает круговой процесс при неизменных температуре и объёме или неизменных температуре и давлении, то алгебраическая сумма тепловых эффектов реакций должна быть равна нулю. В результате кругового процесса значения функций состояния остаются неизменными, а значит алгебраическая сумма тепловых эффектов должна быть равна нулю. Из закона Гесса вытекают очевидные следствия, имеющие практическое значение: 1. Тепловой эффект образования соединения из исходных веществ не зависит от способа, которым это соединение получено. 2. Тепловой эффект разложения какого-либо химического соединения до определённых продуктов равен и противоположен по знаку тепловому эффекту образования этого соединения из тех же продуктов. 3. Разность между тепловыми эффектами превращения двух различных систем в одинаковые продукты реакции равна тепловому эффекту перехода одной системы в другую. Или наоборот: разность тепловых эффектов превращения двух одинаковых химических систем в различные продукты реакции равна тепловому эффекту перехода одних продуктов реакции в другие. Энергия Гиббса Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (Δ H), и энтропийным T Δ S, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж):
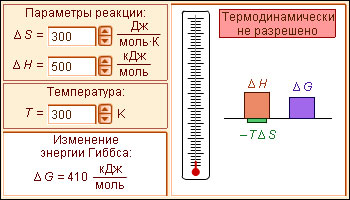
При Δ G < 0 реакция термодинамически разрешена и система стремится к достижению условия Δ G = 0, при котором наступает равновесное состояние обратимого процесса; Δ G > 0 указывает на то, что процесс термодинамически запрещен (рис. 4.4).
Записав уравнение (4.2) в виде Δ H = Δ G + T Δ S, получим, что энтальпия реакции включает свободную энергию Гиббса и «несвободную» энергию Δ S · T. Энергия Гиббса, представляющая собой убыль изобарного (P = const) потенциала, равна максимальной полезной работе. Уменьшаясь с течением химического процесса, Δ G достигает минимума в момент равновесия (Δ G = 0). Второе слагаемое Δ S · T (энтропийный фактор) представляет ту часть энергии системы, которая при данной температуре не может быть превращена в работу. Эта связанная энергия способна лишь рассеиваться в окружающую среду в виде тепла (рост хаотичности системы).
Итак, в химических процессах одновременно изменяются энергетический запас системы (энтальпийный фактор) и степень ее беспорядка (энтропийный фактор, не совершающая работу энергия). Анализ уравнения (4.2) позволяет установить, какой из факторов, составляющих энергию Гиббса, ответственен за направление протекания химической реакции, энтальпийный (Δ H) или энтропийный (Δ S · T). · Если Δ H < 0 и Δ S > 0, то всегда Δ G < 0 и реакция возможна при любой температуре. · Если Δ H > 0 и Δ S < 0, то всегда Δ G > 0, и реакция с поглощением теплоты и уменьшением энтропии невозможна ни при каких условиях. · В остальных случаях (Δ H < 0, Δ S < 0 и Δ H > 0, Δ S > 0) знак Δ G зависит от соотношения Δ H и T Δ S. Реакция возможна, если она сопровождается уменьшением изобарного потенциала; при комнатной температуре, когда значение T невелико, значение T Δ S также невелико, и обычно изменение энтальпии больше T Δ S. Поэтому большинство реакций, протекающих при комнатной температуре, экзотермичны. Чем выше температура, тем больше T Δ S, и даже эндотермические реакции становятся осуществляемыми. КОНЦЕНТРАЦИИ РАСТВОРОВ Важнейшей характеристикой любого раствора является его состав, который выражается концентрацией. Концентрацией раствора называется количество растворенного вещества в определенном массовом или объемном количестве раствора или растворителя. Для приблизительного выражения концентрации растворов используют термины концентрированный и разбавленный растворы. Концентрированный раствор содержит такие количества растворенного вещества, которые сравнимы с количеством растворителя. Например, в 100г воды растворено 20г поваренной соли. Это концентрированный раствор (20 и 100 сравнимые величины). Разбавленный раствор содержит очень малое количество растворенного вещества по сравнению с количеством растворителя. Например, в 100г воды растворено 0,2г поваренной соли. Это разбавленный раствор (0,2г соли очень мало по сравнению со 100г растворителя). Границы между концентрированными и разбавленными растворами условны. Существуют различные способы численного выражения концентрации растворов: массовая доля (%), объемная доля (%), молекулярные и атомные проценты, молярность, нормальность или молярная концентрация эквивалента, моляльность, мольная, атомная и массовые доли, титр и т.д. Важной характеристикой растворов служит их концентрация, которая выражает относительное количество компонентов в растворе. Различают массовые и объемные концентрации, размерные и безразмерные. К безразмерным концентрациям (долям) относятся следующие концентрации:
К размерным концентрациям относятся следующие концентрации:
Массовые концентрации (массовая доля, процентная, моляльная) не зависят от температуры; объемные концентрации относятся к определенной температуре. Диссоциация воды. Водородный показатель. Вода – слабый электролит. Н2О ↔ Н⁺ + ОН ̄ Произведение концентраций ионов водорода и гидроксид-ионов называют ионным произведением воды КН2О. Установлено, что при 25oС ионное произведение воды КН2О = 10 ̄¹⁴. Ионное произведение воды даёт возможность вычислить концентрацию гидроксид-ионов ОН⁻ в любом водном растворе, если известна концентрация ионов водорода Н⁺, и наоборот. Например, если [ОН ̄ ] = 10⁻⁹ моль/л, то [Н⁺] = 10 ̄¹⁴/10⁻⁹ = 10⁻⁵ моль/л Среду любого водного раствора можно охарактеризовать концентрацией ионов водорода Н⁺ или гидроксид-ионов ОН⁻. Нейтральная – это среда, в которой концентрация ионов водорода равна концентрации гидроксид-ионов: [Н⁺] = [ОН ̄ ] = 10⁻⁷ моль/л Кислотная – это среда, в которой концентрация ионов водорода больше концентрации гидроксид-ионов:
[Н⁺] > [ОН ̄ ], [Н⁺] > 10⁻⁷ моль/л Щелочная – это среда, в которой концентрация ионов водорода меньше концентрации гидроксид-ионов: [Н⁺] < [ОН ̄ ], [Н⁺] < 10⁻⁷ моль/л Для характеристики сред растворов удобно Водородным показателем рН называется Например, если [Н⁺] = 10-4 моль/л, то рН = 4, среда раствора кислотная; если [Н⁺] = 10-10 моль/л, то рН = 10, среда раствора щелочная; если [Н⁺] = 10-7 моль/л, то рН = 7, среда нейтральная. Чем меньше pH, тем больше концентрация ионов Н⁺, то есть больше кислотность среды; и наоборот, чем больше рН, тем больше щелочность раствора Гидролиз солей Гидролиз – процесс обменного взаимодействия ионов соли с водой, приводящий к образованию малодиссоциированных веществ и сопровождающийся изменением реакции (pH) среды. Суть гидролиза солей заключается в том, что происходит смещение равновесия диссоциации воды вследствие связывания одного из ее ионов с образованием малодиссоциированного или труднорастворимого вещества. В результате гидролиза могут образовываться молекулы слабых кислот и оснований, анионы кислых солей или катионы основных солей. В большинстве случаев гидролиз является обратимым процессом. При повышении температуры и разбавлении гидролиз усиливается. Гидролиз идет по-разному в зависимости от силы кислоты и основания, образовавших соль. Рассмотрим различные случаи гидролиза солей.
а) Соль образована слабой кислотой и сильным основанием (K2S). При растворении в воде K2S диссоциирует K2S При составлении уравнений гидролиза в первую очередь необходимо определить ионы соли, связывающие ионы воды в малодиссоциирующие соединения, т.е. ионы, обусловливающие гидролиз. В данном случае ионы S2- связывают катион H+, образуя ион HS– S2– +H2O Уравнение гидролиза в молекулярной форме K2S + H2O Практически гидролиз соли преимущественно ограничивается первой ступенью с образованием кислой соли (в данном случае KHS). Таким образом, гидролиз соли, образованной сильным основанием и слабой кислотой (такой, как K2S) протекает по аниону соли. Избыток ионов OH– в растворе обусловливает щелочную реакцию среды в растворе (pН>7).
б) Cоль образована слабым основанием и сильной кислотой (CuCl2, Al2(SO4)3). При растворении в воде CuCl2 диссоциирует СuCl2 Ионы Cu2+ соединяются с ионами OH–, образуя гидроксоионы CuOH+. Гидролиз соли ограничивается первой ступенью, и образование молекулы Cu(OH)2 не происходит. Ионно-молекулярное уравнение имеет вид Cu2+ + HOH В данном случае продуктами гидролиза являются основная соль и кислота. Уравнение гидролиза в молекулярной форме записывается следующим образом CuCl2 + H2O Таким образом, гидролиз соли, образованной слабым основанием и сильной кислотой (в данном случае CuCl2) протекает по катиону соли. Избыток ионов H+ в растворе обусловливает кислую реакцию среды в растворе (рН<7).
При растворении в воде Al2(SO4)3 диссоциирует Al2(SO4)3 В данном случае ионы Al3+ соединяются с ионами ОН-, образуя гидроксоионы AlOH2+. Гидролиз соли ограничивается первой ступенью, и образование молекулы Al(OH)3 не происходит. Ионно-молекулярное уравнение имеет вид Al3+ + Н2О Продуктами электролиза является основная соль и кислота. Уравнение гидролиза в молекулярной форме записывается следующим образом Al2(SO4)3+2 Н2О в) Соль образована слабой кислотой и слабым основанием (CH3COONH4). CH3COO– + NH4+ + H2O В этом случае образуются два малодиссоциированных соединения, и pH раствора зависит от относительной силы кислоты и основания. Если продукты гидролиза могут удаляться из раствора, то гидролиз протекает до конца. Например Al2S3 + 6 H2O = 2Al(OH)3↓ + 3H2S. Возможны и другие случаи необратимого гидролиза, их нетрудно предсказать, ведь для необратимости процесса небходимо, чтобы хотя бы один из продуктов гидролиза уходил из сферы реакции. г) Соли, образованные сильной кислотой и сильным основанием (NaCl, K2SO4, RbBr и др.) гидролизу не подвергаются, т.к. единственным малодиссоциирующим соединением является H2O (рН=7). Растворы этих солей имеют нейтральную среду. Например NaCl + H2O Na+ + Cl– + H2O H2O Реакции обратимого гидролиза полностью подчиняются принципу Ле–Шателье. Поэтому гидролиз соли можно усилить (и даже сделать необратимым) следующими способами: 1) добавить воды; 2) нагреть раствор, при этом усиливается эндотермическая диссоциация воды, а значит, увеличивается количество ионов Н+ и ОН–, которые необходимы для осуществления гидролиза соли; 3) связать один из продуктов гидролиза в труднорастворимое соединение или удалить один из продуктов в газовую фазу; например, гидролиз цианида аммония NH4CN будет значительно усиливаться за счет разложения гидрата аммиака с образованием аммиака NH3 и воды: NH4+ + CN– + H2O Гидролиз можно подавить, действуя следующим образом: 1) увеличить концентрацию растворенного вещества; 2) охладить раствор (для ослабления гидролиза растворы солей следует хранить концентрированными и при низких температурах); 3) ввести в раствор один из продуктов гидролиза; например, подкислять раствор, если его среда в результате гидролиза кислая, или подщелачивать, если щелочная. Взаимное усиление гидролиза Допустим, что в разных сосудах установились равновесия CO32– + H2O Al3+ + H2O Обе соли гидролизованы незначительно, но если растворы смешать, то происходит связывание ионов H+ и OH–. В соответствии с принципом Ле-Шателье оба равновесия смещаются вправо, гидролиз усиливается и протекает полностью 2 AlCl3 + 3 Na2CO3 + 3 H2O = 2 Al(OH)3↓ + 3 CO2 + 6 NaCl. Это называется взаимным усилением гидролиза. Таким образом, если смешивать растворы солей, из которых одна гидролизуется по катиону, а другая – по аниону, гидролиз усиливается и протекает полностью.
7 Скорость химической реакции измеряется изменением количества вещества одного из реагентов или продуктов реакции в единицу времени в единицу объёма для гомогенных систем или на единицу поверхности для гетерогенных систем. Для гомогенных систем, для которых реакция происходит во всем объёме системы, скорость химической реакции определяется следующим уравнением:
V – объём системы
Отношение ± ∆n / V – это молярная концентрация вещества. И тогда ± ∆с. Для гомогенных систем скорость реакции выражается в Для гетерогенных систем, в которых реакция протекает на границе раздела фаз, уравнение для определения скорости химической реакции будет такое:
S – Площадь поверхности раздела фаз, на которой идет химическая реакция. Скорость реакции – величина положительная, поэтому знак ± перед формулой дает возможность выбора. Ставится (+), если скорость реакции определяется по изменению количества продукта реакции. Ставится (-), если скорость реакции определяется по изменению количества исходного вещества. Для гетерогенных систем скорость реакции выражается в Раздел химии, в котором изучаются скорости химических реакций, называется химической кинетикой. Химическая реакция происходит в результате столкновения частиц реагирующих веществ. Но не всякое столкновение частиц приводит к образованию продуктов реакции. Если при столкновении частицы не обладают достаточной энергией, то столкновение будет неэффективным. Такое столкновение называют упругим. Оно подобно столкновению бильярдных шаров. Если энергия частиц будет достаточно высока, то столкновение будет эффективным, и произойдёт химическая реакция. В некоторых случаях (реакция распада) не требуется взаимодействия частиц, но для их прохождения также нужна определённая энергия.
Обратимыми называются реакции, которые могут протекать как в прямом, так и в обратном направлении. Необратимыми называют реакции, протекающие до конца, то есть до тех пор, пока полностью не израсходуется одно из исходных веществ. Необратимые реакции:
Обратимые реакции:
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции
соответствующие константы равновесия выражаются следующим образом:
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений. Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие. Приняв, что V1 = V2, можно записать:
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л
Характер изменения энергии Гиббса позволяет судить о принципиальной возможности осуществления процесса. При ΔG < 0 процесс может протекать, при ΔG > 0 процесс протекать не может (иными словами, если энергия Гиббса в исходном состоянии системы больше, чем в конечном, то процесс принципиально может протекать, если наоборот — то не может). Если же ΔG = 0, то система находится в состоянии химического равновесия. Существует соотношение, связывающее изменение свободной энергии Гиббса в ходе химической реакции с её константой равновесия К: любая реакция может быть рассмотрена как обратимая (даже если на практике она таковой не является). При этом константа равновесия определяется как G = -RT*lnK, К - константа равновесия, процесс идет самопроизвольно при Окислителем называют реагент, который принимает электроны в ходе окислительно-восстановительной реакции. Восстановителем называют реагент, который отдает электроны в ходе окислительно-восстановительной реакции. Окислением называют процесс отдачи электронов атомом, молекулой или ионом, который сопровождается повышением степени окисления. Восстановлением называют процесс присоединения электронов атомом, молекулой или ионом, который сопровождается понижением степени окисления. 1. Если элемент проявляет в соединении высшую степень окисления, то это соединение может быть окислителем. 2. Если элемент проявляет в соединении низшую степени окисления, то это соединение может быть восстановителем. 3. Если элемент проявляет в соединении промежуточную степень окисления, то это соединение может быть как воcстановителем, так и окислителем. Пределы окисления и восстановления элемента выражаются максимальным и минимальным значениями степеней окисления *. В этих крайних состояниях, определяемых положением в таблице Менделеева, элемент имеет возможность проявить только одну функцию – окислителя или восстановителя. Соответственно и вещества, содержащие элементы в этих степенях окисления, являются только окислителями (HNO3, H2SO4, HClO4, KMnO4, K2Cr2O7 и др.) или только восстановителями (NH3, H2S, галогеноводороды, Na2S2O3 и др.). Вещества, содержащие элементы в промежуточных степенях окисления, могут быть как окислителями, так и восстановителями (HClO, H2O2, H2SO3 и др.). Окислительно-восстановительные реакции разделяются на три основных типа: межмолекулярные, внутримолекулярные и реакции диспропорционирования. К первому типу относятся процессы, в которых атомы элемента-окислителя и элемента-восстановителя входят в состав разных молекул (примеры см. в разделе 6.1). Внутримолекулярными называются реакции, в которых окислитель и восстановитель в виде атомов разных элементов находятся в составе одной и той же молекулы. Например, термическое разложениехлората калия по уравнению: 2 KClO3 → 2 KCl + 3 O2 Реакциями диспропорционирования называют процессы, в которых окислителем и восстановителем является один и тот же элемент в одной и той же степени окисления, которая в реакции одновременно как снижается, так и повышается, например: 3 HClO → HClO3 + 2 HCl Возможны также реакции обратного диспропорционирования. К ним относятся внутримолекулярные процессы, в которых окислителем и восстановителем является один и тот же элемент, но в виде атомов, находящихся в разной степени окисления и выравнивающих ее в результате реакции, например: NH4NO2 → N2 + 2 H2O.
Стандартный электродный потенциал — это потенциал металла, определенный относительно стандартного (нормального) водородного электрода, при условии, что концентрация ионов водорода Н+ и ионов испытуемого металла Men+ равны 1 моль-ион/л при стандартных условиях (298К, 101кП). Ряд стандартных электродных потенциалов служит для сравнительной характеристики свойств атомов и ионов металлов в растворе. ЭЛЕКТРОХИМИЧЕСКИЙ РЯД НАПРЯЖЕНИЙ, последовательность расположения электродов в порядке возрастания их стандартных электродных потенциалов (см. Стандартный потенциал). Металлич. электроды в водном р-ре электролита образуют след. электрохимический ряд напряжений: Li, К, Rb, Ba, Sr, Ca, Na, Се, Mg, Be, Al, Ti, Mn, V, Zn, Cr, Ga, Fe, Cd, In, Tl, Co, Ni, Sn, Pb, H2, Bi, Cu, Hg, Ag, Pt, O2, Au. Для сравнения включены водородный электрод (Pt, H2[l атм] | Н+), потенциал к-рого при давлении водорода 1,01 x 105 Па и термодинамич. активности а ионов Н+ в водном р-ре, равной 1, при всех т-рах принимается равным нулю (потенциалопределяющая р-ция Н+ + е6037-30.jpg1/2Н2, где е - электрон) и кислородный электрод (потенциалопределяющая р-ция О2 + 2Н2О + 4е6037-31.jpg 4ОН-). Электрохимический ряд напряжений позволяет судить о термодинамич. возможности протекания тех или иных электродных процессов. Металл с более отрицат. потенциалом может вытеснять металл с менее отрицат. потенциалом из р-ров его солей, растворяясь при этом. Металлы, имеющие отрицат. стандартный потенциал по сравнению с водородным электродом (т. наз. электроотрицат. металлы), в р-рах с не слишком большой термодинамич. активностью ионов металла имеют более отрицат. потенциал, чем водородный электрод в сильно кислых р-рах. Поэтому при замыкании такого электрода с водородным между ними протекает ток, металл растворяется, а на водородном электроде выделяется водород (см. Анодное растворение). Электроотрицат. металлы термодинамически неустойчивы в водных р-рах (их наз. неблагородными металлами) и осаждаются на катоде при более отрицат. потенциале, чем потенциал выделения Н2 (см. Электроосаждение). Металлы, потенциал к-рых менее положительный, чем у кислородного электрода, термодинамически неустойчивы в контакте с О2 (или воздухом) и водой. Поэтому электрохимический ряд напряжений служит для ориентировочных оценок скорости электрохим. коррозии в водных р-рах при обычных т-рах, а также для выбора безопасных контактных пар (гальванич. пар) разнородных металлов. Если металл электроотрицательнее, чем Н2, то может идти активный коррозионный процесс (см. Коррозия металлов, Коррозионностойкие материалы, Электрохимическая защита). Практич. реализация электродных процессов определяется наряду с термодинамич. также и кинетич. факторами (см. Электрохимическая кинетика). Положение в электрохимическом ряду напряжений металлов, образующих ионы разного заряда, зависит от природы соответствующих ионов. Аналогичные ряды напряжений можно построить для неметаллич. и редокс-электродов (окислит.-восстановительных). О. А. Петрий. Уравнение Нернста — уравнение, связывающее окислительно-восстановительный потенциал системы с активностями веществ, входящих вэлектрохимическое уравнение, и стандартными электродными потенциалами окислительно-восстановительных пар. Вывод уравнения Нернста Нернст изучал поведение электролитов при пропускании электрического тока и открыл закон. Закон устанавливает зависимость между электродвижущей силой (разностью потенциалов) и ионной концентрацией. Уравнение Нернста позволяет предсказать максимальный рабочий потенциал, который может быть получен в результате электрохимического взаимодействия, когда известны давление и температура. Таким образом, этот закон связывает термодинамику с электрохимической теорией в области решения проблем, касающихся сильно разбавленных растворов. · · · · · · Если в формулу Нернста подставить числовые значения констант
Гальванический элемент представляет собой электрохимическую систему, состоящую из двух электродов (любого типа), растворы которых соединены с помощью солевого мостика. На рис. 2. представлена схема такого элемента.
Поскольку цинк является металлом имеющим наиболее электроотрицательный потенциал, чем медь (Е0Zn / Zn2+ = -0,76 В, Е0 Cu / Cu2+ = 0,34 В), первое равновесие по сравнению со вторым смещено вправо, следовательно, на цинковомэлектроде имеется избыток электроно
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-07; просмотров: 694; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.136.22.12 (0.02 с.) |

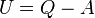 .
.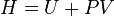 ,
, — давление, а
— давление, а  — объём.
— объём.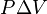 , приращение энтальпии вквазистатическом изобарном процессе равно количеству теплоты, полученному системой.
, приращение энтальпии вквазистатическом изобарном процессе равно количеству теплоты, полученному системой.
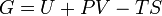
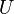 — внутренняя энергия,
— внутренняя энергия,  — давление,
— давление, 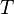 — абсолютна ятемпература,
— абсолютна ятемпература, 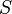 — энтропия.
— энтропия.

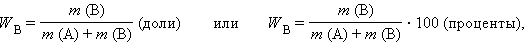

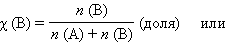
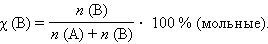

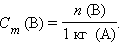


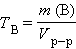 г∙мл–1 или
г∙мл–1 или
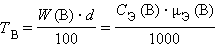 г∙мл–1.
г∙мл–1.
 2K+ + S2-.
2K+ + S2-.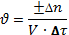
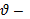 скорость химической реакции
скорость химической реакции – изменение количества вещества
– изменение количества вещества – интервал времени, в котором определяют скорость реакции
– интервал времени, в котором определяют скорость реакции
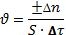






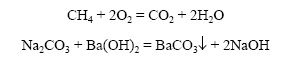
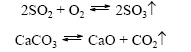

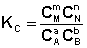 (I.78)
(I.78) 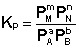 (I.79)
(I.79)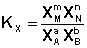 (I.80)
(I.80) (I.81)
(I.81)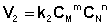 (I.82)
(I.82) (I.83)
(I.83)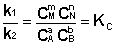 (I.84)
(I.84)

 , где
, где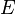 — электродный потенциал,
— электродный потенциал, 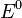 — стандартный электродный потенциал, измеряется в вольтах;
— стандартный электродный потенциал, измеряется в вольтах;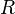 — универсальная газовая постоянная, равная 8.31 Дж/(моль·K);
— универсальная газовая постоянная, равная 8.31 Дж/(моль·K);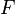 — постоянная Фарадея, равная 96485,35 Кл·моль−1;
— постоянная Фарадея, равная 96485,35 Кл·моль−1; — число электронов, участвующих в процессе;
— число электронов, участвующих в процессе; и
и 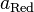 — активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции.
— активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции. получим
получим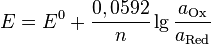
 Рис.2. Гальванический элемент
Даниеля-Якоби
Рис.2. Гальванический элемент
Даниеля-Якоби



