Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Предмет и методы химической термодинамики. Взаимосвязь между процессами обмена веществ и энергии в организме. Химическая термодинамика как теоретическая основа биоэнергетики.Содержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Предмет и методы химической термодинамики. Взаимосвязь между процессами обмена веществ и энергии в организме. Химическая термодинамика как теоретическая основа биоэнергетики. предметом изучения химической термодинамики являются превращения различных видов энергии при протекании химических реакций, процессов растворения, испарения, кристаллизации, адсорбции. Химическая термодинамика количественно определяет тепловые эффекты вышеперечисленных процессов, выясняет возможность самопроизвольного их протекания в том или ином направлении и условия, при которых химические реакции будут находиться в состоянии равновесия. Причем изучение происходящих превращений не требует, с позиций термодинамики, сведений об их механизмах, представлений о строении молекул, участвующих в реакциях веществ. Достаточно только знать начальное состояние системы и те внешние условия, в которых она находится. Следует подчеркнуть также, что термодинамика не может ответить на вопросы о том, с какой скоростью и по какому механизму будет протекать тот или иной процесс, какое количество времени потребуется для достижения в нем химического равновесия. В настоящее время термодинамический метод исследования является одним из наиболее надежных и эффективных средств для изучения обмена веществ и энергии, происходящего в клетках животных, растений и человека. Живые организмы усваивают поступающие к ним из окружающей среды вещества, перерабатывают их, синтезируют и накапливают новые высокомолекулярные соединения для создания и обновления клеток и тканей, аккумулирования больших запасов химической энергии. Совокупность всех процессов называют ассимиляцией или анаболизмом Одновременно в организме протекают противоположные процессы – диссимиляция или катаболизм, сводящиеся к разложению сложных органических соединений, окислению их до Н2О, СО2 и высвобождению при этом энергии. В раннем периоде развития в организме человека, как и во всем живом, процессы ассимиляции превалируют над диссимиляцией, но по мере приближения к старости начинают доминировать процессы диссимиляции, что приводит к уменьшению в организме запасов химической энергии. Изучением и решением всех этих вопросов занимается биоэнергетика, которую можно рассматривать одновременно и как часть биохимии и как часть биофизики. Теоретической же основой биоэнергетики и инструментом, с помощью которого она решает свои задачи, является химическая термодинамика. Основные понятия термодинамики. Внутренняя энергия. Работа и теплота - две формы передачи энергии. Понятия термодинамики:
Система – любой объект природы, состоящий из большого числа молекул (структурных единиц) и отделённый от других объектов природы реальной или воображаемой граничной поверхностью (границей раздела). Состояние системы – совокупность свойств системы, позволяющих определить систему с точки зрения термодинамики. Внутренняя энергия системы – сумма энергий теплового движения молекул и энергии взаимодействия между ними или функция состояния системы, приращение которой равно теплоте Qv. полученной системойв изохорном процессе. Внутреннюю энергию нельзя определить, поскольку у системы нельзя отнять всю энергию. Фаза - гомогенная часть гетерогенной системы (вода и лед в стакане) Фазовый переход - превращения фаз (таяние льда, кипение воды). Функции состояния - свойства, величины которых при переходе системы из одного состояния в другое зависят только от начального и конечного состояния системы и не зависят от пути перехода, получили название функций состояния. К ним относятся, например, давление, объем, температура системы. Термодинамические функции: состояния системы: применяются для определения изменения энергии в тех или иных условиях.(например ΔЕ – внутренняя энергия системы) Изменение ΔЕ обусловлено работой W, которая совершается при взаимодействии системы со средой, и передачей теплоты Q между средой и системой Закон Гесса Тепловой эффект реакции зависит только от природы и состояния исходных вещ-в и не зависит от пути, по которому реакция протекает Следствие 1. Тепловой эффект химической реакции равен разности сумм теплот образования (ΔHf) продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты (ν):
ΔHof,i - стандартная энтальпия образование веществ vi – стехиометрические коэффициенты
Следствие 2. Тепловой эффект химической реакции равен разности сумм теплот сгорания (ΔHc) исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты (ν):
ΔHoс,i- стандартная энтальпия сгорания веществ vi – стехиометрические коэффициенты
Следствие 3. Энтальпия реакции равна разности сумм энергий связей E св исходных и конечных реагентов с учетом их стехиометрических коэффициентов. В ходе химической реакции энергия затрачивается на разрушение связей в исходных веществах (ΣEисх) и выделяется при образовании продуктов реакции (–ΣEпрод). Отсюда
Следовательно, экзотермический эффект реакции свидетельствует о том, что образуются соединения с более прочными связями, чем исходные. В случае эндотермической реакции, наоборот, прочнее исходные вещества. При определении энтальпии реакции по энергиям связей уравнение реакции пишут с помощью структурных формул для удобства определения числа и характера связей.
Следствие 4. Энтальпия реакции образования вещества равна энтальпии реакции разложения его до исходных веществ с обратным знаком.
Следствие 5. Энтальпия гидратации равна разности энтальпий растворения безводной соли (ΔHoраств.б/с)и кристаллогидрата (ΔHoраств.крист)
Классификации реакций, применяющиеся в кинетике: реакции, гомогенные, гетерогенные и микрогетерогенные; реакции простые и сложные (параллельные, последовательные, сопряженные, цепные). Молекулярность элементарного акта реакции. Кинетические уравнения. Порядок реакции. Период полупревращения
Молекулярность реакции – это количество молекул, которые принимают участие единовременно в одном акте столкновений. Молекулярность можно определить, лишь установив механизм реакции. В зависимости от числа реагирующих молекул (частиц), участвующих в элементарном акте, различают одномолекулярные (мономолекулярные), двухмолекулярные, тримолекулярные реакции. К одномолекулярным реакциям типа А→Р относятся процессы распада молекулы на более простые составные части и реакции изомеризации. Двухмолекулярными называются элементарные реакции вида: А+В→Р и 2А→Р (Н2+J2=2HJ,HJ+HJ=H2+J2,CH3COOCH3+H2O=CH3COOH+CH3OHи т.д.). Значительно реже встречаются трехмолекулярные реакции А+2В→Р или 3А→Р. Во всех случаях вид и количество образующихся продуктов реакции не имеет значения, так как молекулярность определяется только числом молекул веществ, реагирующих в элементарном акте. Порядок реакции устанавливается экспериментально. Молекулярность и порядок реакции могут совпадать, а могут и различаться. Молекулярность и порядок реакции совпадают только для простых реакций, протекающих только в одну элементарную стадию без участия посторонних молекул. Молекулярность и порядок реакции не совпадают в трех основных случаях: 1) для сложных реакций; 2) для гетерогенных реакций; 3) для реакций с избытком одного из реагирующих веществ. Период полупревращения – это время, в течение которого прореагирует половина взятого вещества. Кинетическое уравнение выражает зависимость скорости хим. реакции от концентраций компонентов реакционной смеси Молекулярность-число молекул, реагирующих в в одном элементарном химическом акте. Молекулярность реакции определяется числом молекул, вступающих в химическое взаимодействие в элементарном акте реакции. По этому признаку реакции разделяются на мономолекулярные, бимолекулярные и тримолекулярные. Тогда реакции типа А —>В будут являться мономолекулярными, например: а) С16Н34 (t°C) —>CgH18 + С8Н16 - реакция крекинга углеводородов; б) CaC03 (t°C) —>СаО + С02 - термическое разложение карбоната кальция. Тримолекулярные реакции описываются общими уравнениями типа: а) А + В + С Д; б) 2А + В Д; в) 3А Д. Например: а) 2Н2 + 02 2Н20; б) 2NO + Н2N20 + Н20. Скорость реакций в зависимости от молекулярности будет выражаться уравнениями: а) V = к • СА - для мономолекулярной реакции; б) V = к • СА • Св или в) V = к • С2А - для бимолекулярной реакции; г) V = к • С • Св • С э д) V = к • С2А • Св или е) V = k • С3А- для тримолекулярной реакции.
Нередко молекулярность реакции трудно установить, поэтому используют более формальный признак - порядок химической реакции. Порядок реакции равен сумме показателей степеней концентраций в уравнении, выражающем зависимость скорости реакции от концентрации реагирующих веществ (кинетическом уравнении). Порядок реакции чаще всего не совпадает с молекулярностью ввиду того, что механизм реакции, т. е. "элементарный акт" реакции (см. определение признака молекулярности), трудно установить. Рассмотрим ряд примеров, иллюстрирующих указанное положение. 1. Скорость растворения кристаллов описывается уравнениями кинетики нулевого порядка, несмотря на мономолекулярность реакции: AgCl(TB) —>Ag+ + CI", V = k • C(AgCl(TBp= k'C(AgCl(ra}) - p - плотности и является постоянной величиной, т. е. скорость растворения не зависит от количества (концентрации) растворяемого вещества. 2. Реакция гидролиза сахарозы: СО + Н20 —> С6Н1206(глюкоза) + С6Н1206 (фруктоза) является бимолекулярной реакцией, но ее кинетика описывается кинетическим уравнением первого порядка: V=k*Ccax, так как в условиях опытов, в том числе и в организме, концентрация воды есть величина постоянная С(Н20) - const. 3. Реакция разложения водородпероксида, протекающая с участием катализаторов, как неорганических ионов Fe3+, Cu2+ металлической платины, так и биологических - ферментов, например каталазы, имеет общий вид: 2Н202 —> 2Н20 + О э т. е. является бимолекулярной. 13.Зависимость скорости реакции от концентрации. Кинетические уравнения реакций первого, второго и нулевого порядков. Экспериментальные методы определения скорости и константы скорости реакций. Элементарный акт химической реакции осуществляется в момент столкновения реагирующих частиц. Увеличение концентрации реагентов соответствует увеличению числа частиц в объеме, что приводит к более частым столкновениям, а следовательно к увеличению скорости реакции. Количественная зависимость скорости реакции от концентрации выражается законом действующих масс: Скорость простой гомогенной реакции при постоянной температуре пропорциональна произведению концентраций реагирующих веществ, возведенных в степени, численно равные их стехиометрическим коэффициентам.
, где а и б - стехиометрические коэффициенты реагентов, с(А) и с(В) – молярные концентрации реагентов, к- константа скорости реакции. Порядок реакции по реагенту равен показателю степени, в которую надо возвести концентрацию данного реагента в кинетическом уравнении сложной реакции, чтобы вычисленная по этому уравнению скорость была равна скорости, найденной экспериментально. Реакция нулевого порядка Кинетическое уравнение имеет следующий вид:
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ. Нулевой порядок характерен, например, для гетерогенных реакций в том случае, если скорость диффузии реагентов к поверхности раздела фаз меньше скорости их химического превращения. Реакция первого порядка Кинетическое уравнение реакции первого порядка:
Приведение уравнения к линейному виду даёт уравнение:
Реакция второго порядка Для реакций второго порядка кинетическое уравнение имеет следующий вид:
или
Измерение скорости реакции основано на определении концентрации одного из реагирующих веществ через различные промежутки времени от начала реакции. Для определения концентраций можно применять методы физико-химического анализа, основанные на зависимости физических свойств смеси от её состава (например, определение показателя преломления, угла вращения плоскости поляризации, вязкости, электрической проводимости, объёма, плотности, изменения температур замерзания и кипения, интенсивности окраски и т. п.), и методы аналитической химии (например, титрование). Поскольку концентрации по ходу реакции непрерывно меняются, то необходимо или очень быстрое измерение концентрации (методы физико-химического анализа), или торможение реакции во взятой пробе (химический контроль). Торможение может быть достигнуто охлаждением, резким разбавлением, устранением катализатора или совместным действием всех указанных факторов. Если реакция, протекающая в газовой фазе, сопровождается изменением числа молекул, то её течение удобно контролировать по изменению давления смеси во времени. К сравнительно медленным реакциям со временем полупревращения порядка получаса и более можно применять спектроскопию, масс-спектрометрию и хроматографию. Для исследования скоростей очень быстрых реакций (с периодом полупревращения до 10-7и даже 10–9с) используются специально разработанные методы и особая аппаратура. Для определения порядка реакции необходимо иметь экспериментальные данные об изменении концентрации реагирующих веществ со временем. Если в реакции участвует несколько веществ, то пользуются методом изолирования Оствальда. 14.Зависимость скорости реакции от температуры. Правило Вант - Гоффа. Температурный коэффициент скорости реакции и его особенности для биохимических процессов. Правило Вант-Гоффа Или (учебник Слесарев): Энергия активации
Уравнение Аррениуса Чем больше энергия активации, тем меньше будут константа и скорость химической реакции, так как в системе будет меньше число активных частиц. Стерический фактор (P) Первый закон Рауля Пар, находящийся в равновесии с жидкостью, называют насыщенным. Давление такого пара над чистым растворителем (p0) называют давлением или упругостью насыщенного пара чистого растворителя. Франсуа Мари Рауль: "Давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально мольной доле растворителя в данном растворе: p = p0 · χр-ль, где p — давление пара над раствором, Па; p0 — давление пара над чистым растворителем; χр-ль —— мольная доля растворителя. 1. Понижение давления пара над раствором Это свойство мы уже рассмотрели для случая, когда растворенное вещество является летучим (закон Рауля). При растворении нелетучего вещества давлением пара растворенного вещества можно пренебречь и учитывать лишь давление пара растворителя Р0. Давление пара растворителя над раствором нелетучего вещества может быть выражено уравнением: Р = Р0 · N1, Вместо N1 введем мольную долю растворенного вещества N2. Учитывая, что N1+ N2 = 1, получим Р = Р0 · (1- N2).
Второй закон Рауля: Повышение температуры кипения или понижение температуры замерзания идеальных растворов нелетучих веществ прямо пропорционально моляльной концентрации раствора:
Δ Т кип = К кип * С m Δ Ткр = К кр * Сm
Здесь Cm -моляльная концентрация раствора (моль/кг); Ккип - эбуллиосконическая константа или константа кипения растворителя; Ккр - криоскопическая константа или константа кристаллизации растворителя. Растворение нелетучего вещества в растворителе приводит к расширению температурного диапазона существования жилкой фазы: раствор замерзает при более низкой температуре, а кипит при более высокой температуре по сравнению с растворителем, т.к. моляльная концентрация раствора повышается. Экспериментально определенное понижение температуры замерзания плазмы крови человека равно 0,56 °С, что отвечает моляльной в молярной концентрациям частиц 0,303 моль/кг или 0,303 моль/л и совпадает с величиной, полученной при измерении осмотических показателей крови. Все коллигативные свойства растворов находятся в тесной взаимосвязи. Анализ коллигативных свойств водных растворов убедительно свидетельствует о том, что растворенные вещества сильно влияют на свойства воды как растворителя, а раствор является системой, свойства которой резко отличаются от свойств исходных компонентов. Это очень важно понимать при изучении особенностей биожидкостей организма, которые содержат воду, электролиты, белки, углеводы и вещества в коллоидном состоянии. В то же время физико-химические свойства таких систем, а особенно биологические и физиологические функции, определяются не только качественным и количественным составом, но и структурой систем, которая динамична и еще недостаточно изучена.
Второй закон Рауля(следствия) – 1) условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твердым растворителем. Это будет достигаться только при более низких температурах, чем температура замерзания растворителя. 2 ) жидкость кипит при той температуре, при которой общее давление насыщенного пара становится равным внешнему давлению. Если вещество нелетучее, то давление должно быть равным парциальному давлению растворителя. 2. Понижение температуры замерзания растворов Раствор, в отличие от чистой жидкости, не отвердевает целиком при постоянной температуре; при некоторой температуре, называемой Рассмотрим p – T диаграмму состояния растворителя и растворов различной концентрации (рисунок 1), на которой кривая OF – есть зависимость давления пара над твердым растворителем, а кривые OA, BC, DE – зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями, соответственно. Кристаллы растворителя будут находиться в равновесии с раствором только тогда, когда давление насыщенного пара над кристаллами и над раствором одинаково.Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, температура, отвечающая этому условию, всегда будет более низкой, чем температура замерзания чистого растворителя. При этом понижение температуры замерзания раствора ΔTзам не зависит от природы растворенного вещества и определяется лишь соотношениемчисла частиц растворителяи растворенного вещества. Показано, что понижение температуры замерзания раствора ΔTзам, прямо пропорционально моляльной концентрации раствора: ΔTзам, = Kзам · Cm, где Kзам – криоскопическая постоянная растворителя – определяется природой растворителя. Связь криоскопической константы с теплотой плавления растворителя Криоскопическая константа Kзам представляет собой понижение температуры замерзания раствора при растворении 1 моль нелетучего неэлектролита в 1000 г растворителя. Константа Kзам связана с температурой замерзания растворителя Т и его теплотой плавления ΔНпл уравнением: Kзам =(RT2зам/ ΔHпл) · (M1/1000), где M1 – молярная масса растворителя, ΔHпл - энтальпия плавления растворителя, Т зам –температура замерзания растворителя.Для данного растворителя криоскопическая постоянная не зависит от природырастворенного вещества. Kзам = (RT2зам / ΔHпл) · (M1 / 1000) Метод исследования, основанный на измерении понижения температуры затвердевания растворов называют криоскопическим методом. Зная массу растворителя (g1) и массу растворенного вещества (g2) можно по измеренной ΔTзам, пользуясь уравнением ΔTзам = Kзам·Cm, определить молярную массу растворенного вещества (М2): M2 = Kзам · (1000·g2)/(ΔTзам·g1) 3. Повышение температуры кипения растворов Температура кипения растворов нелетучего вещества всегда выше, чем температура кипения чистого растворителя при том же давлении. Любая жидкость – растворитель или раствор – кипит при той температуре, при которой давление насыщенного пара становится равным внешнему давлению. Соответственно, температуры, при которых изобара p = 1 атм пересечёт кривые, представляющие зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями, соответственно, будут температурами кипения этих жидкостей. Повышение температуры кипения растворов нелетучих веществ ΔTкип = Tкип – T0 кип пропорционально понижению давления насыщенного пара и, следовательно, прямо пропорционально моляльной концентрации раствора: ΔTкип = Kкип · Сm Коэффициент пропорциональности Kкип – есть эбулиоскопическая постоянная растворителя, не зависящая от природырастворенного вещества: Эбулиоскопическая константа имеет физический смысл повышения температуры кипения растворов с моляльной концентрацией, равной 1 моль/кг. Таким образом, второй закон Рауля можно в наиболее общем виде сформулировать следующим образом: «Понижение температуры замерзания и повышение температуры кипения разбавленного раствора нелетучего неэлектролита прямо пропорционально моляльной концентрации раствора и не зависит от природы растворенного вещества». Связь эбулиоскопической константы с теплотой испарения растворителя Kкип = (RT2 кип/ ΔHисп) · (M1/1000) Для данного растворителя эбулиоскопическая постоянная не зависит от природы растворенного вещества. Метод исследования, основанный на измерении повышения температуры кипения растворов, называют эбулиоскопическим методом. Зная массу растворителя (g1) и массу растворенного вещества (g2) можно по измеренной ΔTкип, пользуясь уравнением ΔTкип = Kкип·Cm, определить молекулярную массу растворенного вещества (М2): M2 = Kкип · (1000·g2)/(ΔTкип·g1)
22. Осмос. Осмотическое давление: закон Вант-Гоффа. Осмотическое давление в растворах неэлектролитов и электролитов. Изотонический коэффициент. Осмос - самопроизвольная диффузия молекул растворителя сквозь мембрану с избирательной проницаемостью. Не зависит от природы растворенного вещества, а только от числа частиц в растворе и от температуры. Осмотическое давление - избыточное гидростатическое давление, возникающее в результате осмоса и приводящее к выравниванию скоростей взаимного проникновения молекул растворителя сквозь мембрану с избирательной проницаемостью. Для учета межмолекулярных взаимодействий в реальных растворах было предложено использовать изотонический коэффициент, который для растворов неэлектролитовi=1, а для растворов электролитов i> 1(максимальное значение равно числу ионов в его молекуле) В. Пфеффер и Я Вант-Гофф, изучая колличественную зависимость осмотического давления от внешних факторов, установили, что оно подчиняется объединенному газовому закону Менделеева-Клайперока. Закон Вант-Гоффа гласит: "Осмотическое давление разбавленного раствора равно давлению, которое проявляло бы растворенное вещество, если бы оно было газообразным и занимало объем, равный объему раствора". Уравнение для описания осмотического давления для растворов неэлектролитов можно записать так: р = (m/MV)*RT = C*RT., где р - осмотическое давление, кПа: С - молярная концентрация, моль/ л; R - универсальная газовая постоянная, равная 8,31 (кПа-л)(моль-К); Т - абсолютная температура. К. Концентрация кинетически самостоятельных частиц в растворах электролитов всегда больше, чем это следует из аналитической концентрации. С тем, чтобы свойства растворов электролитов удовлетворительно описывались уравнениями, выражающими следствие из закона Рауля, Вант-Гоффом был введен поправочный эмпирический коэффициент, называемый сейчас изотоническим или коэффициентом Вант-Гоффа (i)
i — изотонический коэффициент раствора; C — молярная концентрация раствора, выраженная через комбинацию основных единиц СИ, то есть, в моль/м3, а не в привычных моль/л; R — универсальная газовая постоянная; T — термодинамическая температура раствора.
. Коллигативные свойства разбавленных растворов электролитов. Элементы теории растворов сильных электролитов Дебая- Хюккеля. Ионная сила, ее математическое выражение. Понятие об активности. Коэффициент активности. Коллигативные свойства – не зависящие от природы частиц свойства, а зависящие только от концентрации частиц в растворе. Такими свойствами являются: диффузия, осмотическое давление, понижение давления насыщенного пара растворителя над раствором, повышение температуры кипения и понижение температуры замерзания раствора. Теория Дебая – Хюккеля. · В растворах сильных электролитов каждый нон окружен со всех сторон ионами противоположенного знака, вследствие чет движение ионов ограничено. · Ионы сильного электролита в растворе взаимодействуют между собой благодаря наличию значительных электростатических сил, в результате чего ноны одного знака образуют вокруг нона другого знака так называемую ионную атмосферу. Необходимо также учитывать сольватацию ионов, · Ионная атмосфера и сольватная оболочка замедляют движение ионов в растворе и являются причиной кажущейся неполной ионизации. · Кроме того, в растворах сильных электролитов при высоких концентрациях может происходить ассоциация ионов. · Для учета этих влиянии состав растворов электролитов следует характеризовать не аналитической, а эффективной концентрацией, называемой активностью (Льюис, 1907),
Основная ее идея - вследствие электростатического притяжения между положительными и отрицательными ионами вблизи каждого иона находятся главным образом ионы противоположного знака, т.е. ион как бы окружен ионной атмосферой. Суммарный заряд этой атмосферы по абсолютной величине равен заряду центрального иона, но противоположен ему по знаку. Тормозящее действие ионной атмосферы на передвижение ионов проявляется таким образом, что все свойства, зависящие от концентрации ионов (такие, как электрическая проводимость, осмотическое давление и т.д.), отвечают заниженной степени диссоциации – кажущейся степени диссоциации. Для оценки состояния ионов в растворе пользуются понятием активности иона – его условной концентрации, соответственно которой он действует при химических реакциях: a = f*C, где a – активность иона, C – его концентрация, f – коэффициент активности. Значение f < 1 указывает на связывающее взаимодействие ионов; если f близок к единице, это говорит о слабом межионном взаимодействии. В очень разбавленных растворах действие межионных сил почти не проявляется. Ионная сила раствора (I) - величина, характеризующая интенсивность электростатического ноля всех ионов в растворе, которая равна полу-сумме произведений молярной концентрации (с) каждого иона на квадрат его заряда (z):
Активность электролита – эффективная концентрация в соответствии с которой он участвует в различных процессах В разбавленных растворах сильных электролитов с одинаковой ионной силой коэффициенты активности катионов и анионов одинаковой зарядности равны независимо от их химической природы. Электролиты в организме.Осмоляльность и осмолярность биологических жидкостей и перфузионных растворов.Понятия изо-, гипо-, гипертонический раствор. Понятие об изоосмии.Роль осмоса и осмотического давления в биологических системах. Плазмолиз. Цитолиз. В биологических системах широко распространены межионные взаимодействия, которые сильно зависят от ионной силы растворов, что прежде всего сказывается на значениях констант диссоциации ионогенных групп биологических субстратов, так как они определяются активностями ионов, а не их концентрациями. Незначительное увеличение ионной силы раствора вызываем изменение степени ионизованности белков или нуклеиновых кислот, вследствие чего
|
|||
|
|
Последнее изменение этой страницы: 2016-04-07; просмотров: 2722; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.214 (0.018 с.) |

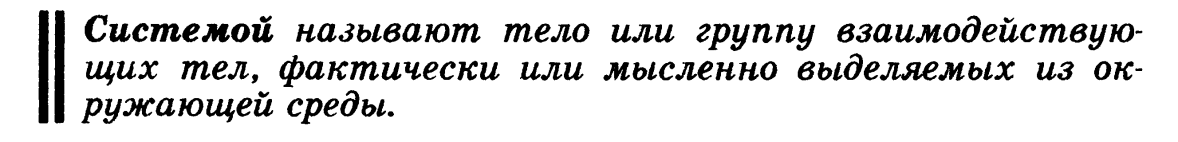















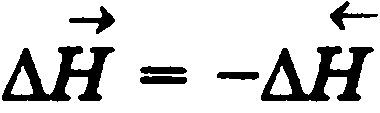





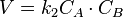

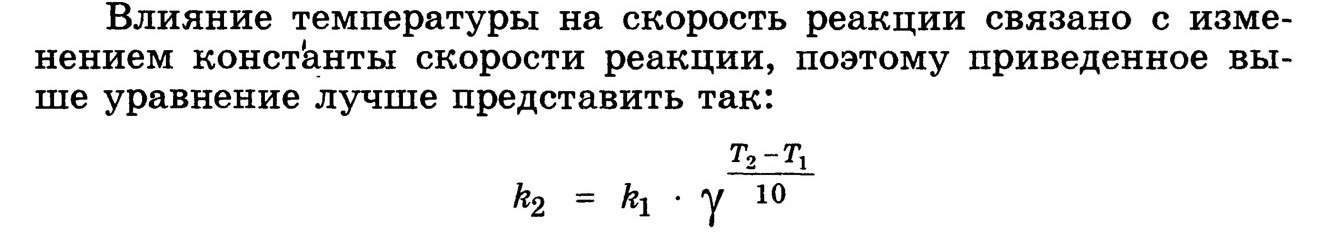
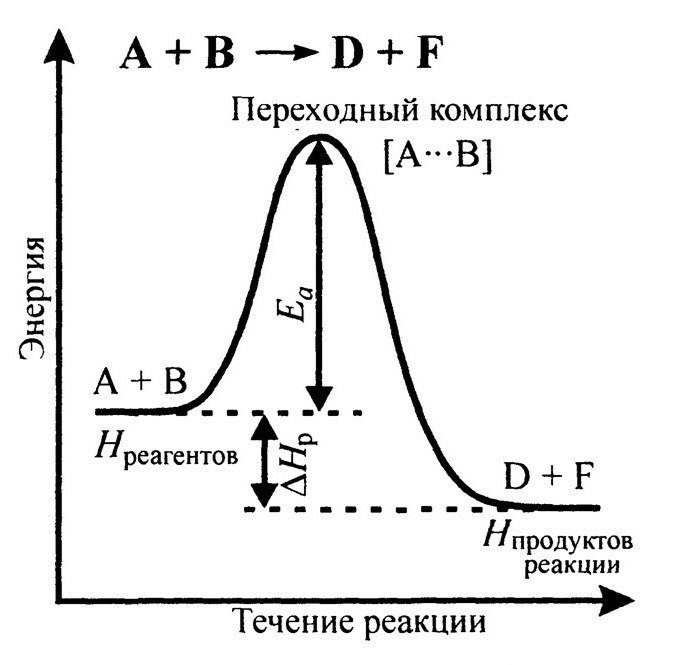
 Энергетический профиль течения реакции
Энергетический профиль течения реакции