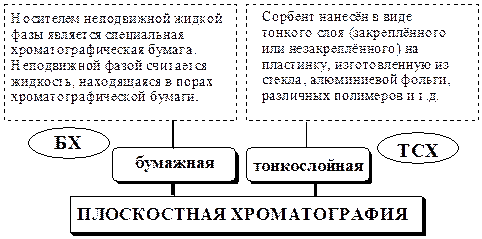Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Природа и свойства электромагнитного излученияСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
Природа и свойства электромагнитного излучения Электромагнитное излучение имеет двойственную природу и обладает как волновыми, так и корпускулярными (дискретными) свойствами. Электромагнитная волна состоит из двух компонентов - электрического и магнитного, которые перпендикулярны друг другу и к направлению движения волны (рис.19.1). В отличие от других волновых процессов, например, звуковых волн для распространения электромагнитного излучения не нужна проводящая среда
Рис. 19.1. Электромагнитная волна Электромагнитная волна, как и любая волна, обладает следующими основными параметрами. Длина волны (l) - расстояние, которое проходит волна за один период её колебаний (расстояние между двумя последовательными максимумами). Длина волны измеряется в метрах (м). На практике обычно используют кратные единицы - нанометр (1 нм = 1×10-9 м) или микрометр (1 мкм = 1×10-6 м). Частота (n)- число колебаний в 1 секунду. Частота измеряется в герцах (1Гц = 1 с-1) или в кратных ему единицах, например, 1МГц = 1×106 Гц. Длина волны и частота колебаний связаны между собой следующим уравнением
где с - скорость распространения волны в данной среде. Для электромагнитной волны
где с0 - скорость света в вакууме (2,99792×108 м/с), n - показатель преломления среды. Частота является более фундаментальной характеристикой, чем длина волны. Она зависит только от свойств источника излучения и не зависит от свойств среды. Длина волны зависит от природы среды, температуры и давления. Волновое число Электромагнитное излучение можно рассматривать как поток частиц энергии - фотонов. Связь между волновой и корпускулярной природой электромагнитного излучения устанавливает уравнение Планка:
где h - постоянная Планка (h = 6,6262×10-34 Дж×с) Единицей измерения энергии является Джоуль (Дж). В спектроскопии часто используют внесистемную единицу - электрон-вольт (1эВ = 1,6022×10-19 Дж). Чем больше длина волны электромагнитного излучения (меньше частота колебаний), тем меньше его энергия. Совокупность всех энергий (длин волн, частот) электромагнитного излучения называется электромагнитным спектром. В спектроскопических методах анализа спектром (спектром поглощения, спектром испускания) называется зависимость между энергией кванта и числом квантов, обладающих данной энергией.
Абсорбционные спектроскопические методы анализа Практическое применение ААС используется для количественного определения более 70 элементов, главным образом, металлов. Зависимость между степенью поглощения электромагнитного излучения и концентрацией поглощающего вещества в ААС такая же, как и в других абсорбционных методах анализа. Оптическая плотность прямо пропорциональна концентрации атомов соответствующего элемента в атомизаторе и, следовательно, в анализируемой пробе
Коэффициент k является эмпирической величиной. Определение концентрации в ААС проводят методом градуировочного графика или методом добавок. В Республике Беларусь ААС используется для определения неорганических токсикантов («металлических ядов») в различных биологических объектах при судебно-химическом и химико-токсикологическом исследовании. Пределы обнаружения большинства элементов составляют при использовании пламени 1-100 мкг/л, при электротермической атомизации на несколько порядков меньше. Основные ограничения данного метода анализа связаны с необходимостью наличия большого набора соответствующих ламп, а также необходимостью переведения анализируемого объекта в раствор. 72 Прямая фотометрия Используется для определения веществ, обладающих достаточно интенсивным собственным поглощением. В прямой фотометрии измеряют оптическую плотность раствора вещества при длине волны, соответствующей максимальному поглощению, и далее одним из способов определяют концентрацию вещества в этом растворе. Прямая фотометрия обычно используется для анализа матриц относительно простого состава, в которых отсутствуют вещества, обладающие таким же характером поглощения, что и определяемое вещество, либо мешающие компоненты можно легко отделить в процессе пробоподготовки. Фотометрические реакции Значительно чаще в фотометрии, особенно в случае определения неорганических веществ, обладающих незначительным собственным поглощением, измерению оптической плотности предшествует проведение химической реакции, в которой образуется новое вещество, обладающее более интенсивным поглощением. Например Fe3+ + nSCN- ® [Fe(SCN)n]3-n В основе получения окрашенных продуктов могут лежать реакции комплексообразования (в том числе и с органическими реагентами), окислительно-восстановительные реакции, различные реакции с участием функциональных групп органических соединений и т.д. К фотометрическим реакциям предъявляются требования: · чувствительность - реакция считается высокочувствительной, если величина кажущегося молярного коэффициента поглощения превышает 6×104 · контрастность - разность между длинами волн, соответствующим максимумам поглощения реагента и продукта реакции должна быть как можно больше; реакция считается высококонтрастной, если Dl > 80 нм. · надёжность - независимость протекания реакции от незначительных изменений условий её проведения, а также от присутствия в растворе других веществ · избирательность - в реакцию должно вступать только определяемое вещество или, по крайней мере, незначительная группа веществ. Иногда проведение фотометрической реакции совмещается с экстракцией образующегося продукта несмешивающимся с водой растворителем. Такой гибридный метод анализа называется экстракционной фотометрией. Экстракционную фотометрию используют в тех случаях, когда продукт фотометрической реакции оказывается мало растворимым в воде или определению мешают другие вещества (либо избыток реагента), присутствующие в растворе.
Фотометрическое титрование Фотометрическим титрованием называется группа титриметрических методов анализа, в которых конечную точку титрования обнаруживают по изменению оптической плотности раствора. В основе фотометрического титрования могут лежать любые реакции, применяемые в титриметрии. Определение может проводиться как без индикатора (если хотя бы один из компонентов используемой реакции способен поглощать электромагнитное излучение выбранного диапазона), так и в присутствии индикаторов. На рис. 20.15 показаны различные варианты кривых фотометрического титрования.
Рис. 20.15. Различные варианты кривых фотометрического титрования 1 - поглощает определяемое вещество, 2 - поглощает титрант, 3 – поглощает продукт реакции, 4 – поглощают и определяемое вещество и титрант Фотометрическое титрование, в отличие от титрования с визуальным обнаружением конечной точки, может быть использовано для анализа разбавленных, окрашенных, мутных растворов, а также в том случае, когда изменение окраски раствора в конечной точке титрования плохо воспринимается глазом. 74 продолжение ИК-спектроскопия К инфракрасному относят электромагнитное излучение с длинами волн примерно от 800 нм (0,8 мкм) до 103 мкм.
С точки зрения использования в анализе наиболее полезной является средняя область ИК-диапазона. Практическое применение ИК-спектроскопия используется преимущественно для установления строения и идентификации органических (реже неорганических) соединений, в том числе и лекарственных веществ. В плане качественного анализа ИК-спектры являются значительно более информативными, чем спектры поглощения в УФ- или видимой области. Большинство функциональных групп (OH-, NH2 и т.п.) не обладают собственным поглощением в УФ- и видимой области. Напротив, в ИК-спектрах они имеют собственные полосы поглощения. Кроме того, в УФ-спектре отдельные полосы поглощения часто сливаются друг с другом, что затрудняет его интерпретацию. Обнаружение и идентификация веществ методом ИК-спектроскопии может проводиться следующим образом: · обнаружение отдельных функциональных групп по характеристическим полосам поглощения, · сравнение ИК-спектров исследуемого соединения и стандартного образца, · идентификация неизвестного соединения с помощью атласа или компьютерной библиотеки ИК-спектров. В количественном анализе ИК-спектроскопия используется значительно реже, чем спектроскопия в УФ- и видимой области. Это связано с тем, что чувствительность данного метода анализа существенно ниже (величины e обычно составляют 1-1×103), а воспроизводимость хуже. Количественный анализ, как и в других абсорбционных спектроскопических методах, основан на законе Бугера-Ламберта-Бера. Концентрацию вещества определяют методом градуировочного графика. 76 Атомно-эмиссионная спектроскопия (АЭС) - спектроскопический метод анализа, основанный на измерении электромагнитного излучения оптического диапазона, испускаемого термически возбуждёнными свободными атомами или одноатомными ионами. Практическое применение АЭС используется для обнаружения и количественного определения различных элементов, обычно металлов. В качественном анализе используется наличие характерных линий в получаемых спектрах испускания. Наиболее подходящий атомизатор для качественного анализа - дуговой разряд, так как пламя даёт спектры бедные спектральные линиями, атомизатор с ИСП - наоборот, очень сложные спектры, которые можно расшифровать только с помощью компьютера. Количественный анализ в АЭС основан на зависимости интенсивности испускания от концентрации данного элемента в анализируемой пробе. Зависимость между интенсивностью спектральных линий и концентрацией элемента в пробе является более сложной, чем, например, в ААС, и описывается уравнением Ломакина-Шайбе
где a и b - эмпирические константы, которые характеризуют процессы, происходящие на поверхности электродов (a) и самопоглощение излучения (b). Зависимость I от C не является линейной (в отличие от зависимости lgI от lgC). Самый большой диапазон линейности наблюдается при использовании атомизатора с ИСП. Для определения концентрации в АЭС применяют метод градуировочного графика и метод добавок. Для построения градуировочного графика часто используют внутренние стандарты. Предел обнаружения в АЭС при определении хорошо атомизируемых и легковозбудимых элементов с использованием пламенного атомизатора составляет 10-7-10-2%, других элементов (ИСП-атомизатор) - 10-8 - 10-2%. Воспроизводимость при использовании пламени и ИСП - Sr = 0,01-0,05, при использовании искры и дуги Sr = 0,05 - 0,2. 77 Люминесцентной спектроскопией называют группу эмиссионных спектроскопических методов анализа, основанных на явлении люминесценции. Люминесценцией называется свечение атомов, молекул и других более сложных частиц, возникающее в результате электронного перехода при их возвращении из возбуждённого состояния в основное. Люминесценцию иногда называют холодным светом, так как обычно температура люминесцирующего тела не отличается от температуры окружающей среды. Природа вещества Неорганические соединения (за исключением некоторых соединений урана, лантанидов) обычно не способны флуоресцировать в растворе. В то же время среди органических соединений флуоресцирующих веществ достаточно много. Необходимым (но не достаточным!) условием для фотолюминесценции является способность вещества поглощать электромагнитное излучение УФ- или видимого диапазона. Обычно вещества, обладающие интенсивной флуоресценцией, имеют длинную систему сопряжённых связей. Наиболее часто флуоресцирующие вещества встречаются среди ароматических соединений. Введение в бензольное кольцо электронодонорных заместителей увеличивает способность вещества флуоресцировать. Например, многие фенолы и ароматические амины обладают интенсивной флуоресценцией. Введение электроноакцепторных заместителей, за некоторым исключением, уменьшает флуоресценцию. Атомы тяжёлых галогенов (Br, I) увеличивают скорость интеркомбинационной конверсии и, тем самым, уменьшают квантовый выход флуоресценции. Однако введение тяжёлых галогенов увеличивает способность вещества фосфоресцировать. Способность вещества к флуоресценции в растворе увеличивается при конденсации ароматических колец и увеличении «жёсткости» молекулы. Например
Концентрация вещества Зависимость между интенсивностью флуоресценции и концентрацией флуоресцирующего вещества в растворе более сложная, чем между поглощением света и концентрацией. Это связано с тем, что процесс излучения является вторичным и зависит от предшествующего ему процесса поглощения света. Рассмотрим простейший случай, когда в растворе находится только одно флуоресцирующее вещество.
Таким образом:
Следовательно, зависимость между интенсивностью флуоресценции и концентрацией флуоресцирующего вещества не является линейной. Функцию
Если произведение
Таким образом, при малых значениях оптической плотности (при lвозб) зависимость интенсивности флуоресценции от концентрации можно считать линейной, что и используется в количественном анализе. При более высоких значениях A зависимость интенсивности флуоресценции от концентрации становится более сложной и отклоняется от линейной. При A = 0,01 отклонение от линейности составляет 1%, 0,05 - 5%; 0,5 - около 35% (рис. 21.4).
Рис. 21.4. Зависимость между интенсивностью флуоресценции и оптической плотностью раствора 1) рассчитанная по упрощённой формуле I = KC; 2) реальная Влияние оптической плотности раствора на интенсивность флуоресценции называется «эффектом внутреннего фильтра». Этот эффект обусловлен двумя причинами: · поглощением возбуждающего света, вследствие чего частицы, находящиеся дальше от источника излучения, будут получать меньше возбуждающего излучения; · поглощением одними частицами вещества излучения, испускаемого другими частицами этого же вещества. ГЛАВА 22
Общая характеристика Хроматография - метод разделения смесей веществ или частиц, основанный на различии в скоростях их перемещения в системе, состоящей из несмешивающихся и движущихся друг относительно друга фаз. Хроматография - гибридный метод анализа, включающий разделение веществ и их последующее определение при помощи специальных устройств - детекторов. В качестве неподвижной фазы в хроматографическом процессе выступает твёрдое вещество (сорбент) или плёнка жидкости, нанесённая на твёрдый носитель, а в качестве подвижной фазы - жидкость или газ, протекающий через неподвижную фазу. В отличие от статических методов разделения - сорбции и экстракции хроматография является динамическим процессом. При перемещении через неподвижную фазу подвижная фаза встречает на своём пути всё новые и новые слои сорбента, что сопровождается многократными повторениями актов сорбции и десорбции разделяемых веществ. Хроматографическое разделение обладает большей эффективностью по сравнению со статическими методами.
Табл. 22.1. Классификация хроматографических методов в зависимости от преобладающего процесса, лежащего в основе разделения веществ
По способу получения хроматограммы хроматография бывает элюентной, фронтальной и вытеснительной (табл 22.2). Табл. 22.2. Способы получения хроматограммы
Табл. 22.3. Приёмы количественного определения в хроматографии
Внутренний стандарт представляет собой специально добавляемое к анализируемой пробе в точно измеренном количестве вещество, свойства которого близки к свойствам определяемого вещества. Вещество, взятое в качестве внутреннего стандарта: · не должно химически взаимодействовать с компонентами анализируемой смеси, с неподвижной или подвижной фазой; · должно отсутствовать в исходной анализируемой смеси; · давать пик, находящийся на хроматограмме в непосредственной близости от пиков определяемых веществ, но не накладывающийся на них и на пики других соединений. Концентрацию внутреннего стандарта выбирают таким образом, чтобы высота (площадь) пика внутреннего стандарта была соизмерима с высотой (площадью) пика определяемого вещества (рис. 22.3).
Рис. 22.3. Хроматограмма, полученная при ГЖХ-определении аминазина в печени (внутренний стандарт – бромгексин) В методе внешнего и внутреннего стандарта используются одни и те же приёмы расчёта содержания вещества (метод градуировочного графика и др.). Основное различие заключается в характере используемого аналитического сигнала. Метод внутреннего стандарта обладает большей надёжностью и даёт более воспроизводимые результаты, особенно в случае сложной пробоподготовки. ГЛАВА 23
Общая характеристика Газовая хроматография - группа хроматографических методов, в которых подвижная фаза газообразна (находится в состоянии газа или пара). В зависимости от агрегатного состояния неподвижной фазы:
Способы ввода пробы Устройство для ввода пробы (дозатор) предназначено для ввода в колонку определённого количества анализируемой пробы. Для дозирования газообразных веществ применяют газовые краны-дозаторы. Если анализируемая проба является жидкостью, её вводят с помощью специального микрошприца в испаритель. Испаритель представляет собой металлический блок, нагреваемый до определённой температуры, имеющий канал для ввода и испарения жидкой пробы. С одной стороны канал закрыт пробкой из самоуплотняющейся термостойкой силиконовой резины, а с другой стороны к нему присоединена хроматографическая колонка. В канал подаётся поток предварительно нагретого газа-носителя. Проба, введённая в канал испарителя, быстро испаряется и переносится потоком газа-носителя в колонку. Температура испарителя обычно выбирается равной или на 30-50 °С более высокой, чем температура кипения наиболее высококипящего компонента анализируемой смеси и, как правило, на 20-30 °С превышает температуру колонки. Хроматографическая колонка В газовой хроматографии применяют колонки двух типов:
Капиллярные колонки обеспечивают более высокую эффективность хроматографического разделения, чем насадочные. Вариант газовой хроматографии, в котором используются капиллярные колонки, называется капиллярной газовой хроматографией. Детекторы Детектор представляет собой устройство, предназначенное для обнаружения и количественного определения компонентов анализируемой смеси, выходящих из колонки в потоке газа-носителя. Работа детектора основана на преобразовании в электрический сигнал изменений физических, химических или физико-химических свойств газового потока, выходящего из колонки. Основными характеристиками хроматографических детекторов являются: · чувствительность, · предел обнаружения, · величина линейного динамического диапазона, · быстродействие, · селективность. Для газовой хроматографии предложено более 50 различных детекторов. Однако обычно комплект современного универсального хроматографа включает в себя не более 4 - 6 детекторов. Основные характеристики некоторых детекторов, применяемых в газовой хроматографии, приведены в табл. 23.1. Табл. 23.1 Характеристика некоторых газохроматографических детекторов
Детектор по теплопроводности (катарометр) представляет собой металлический блок, в полости которого находится тонкая спираль, изготовленная из материала (W, Pt), электрическое сопротивление которого сильно зависит от температуры. Обычно катарометр имеет две ячейки. Через ячейку сравнения пропускают газ-носитель, а через ячейку измерения - элюат. Если через обе ячейки катарометра протекает чистый газ-носитель, теплопроводность среды в них одинакова. Обе спирали имеют одинаковую температуру и одинаковое сопротивление. Если из хроматографической колонки выходит вещество, теплопроводность которого отличается от теплопроводности газа-носителя, то температура и сопротивление спирали, находящейся в измерительной ячейке, изменяются. Различие сопротивлений спиралей определяется с помощью моста Уитстона (рис. 23.3). При использовании катарометра в хроматографе должны быть две колонки, через одну пропускают газовую смесь, содержащую разделяемые вещества, а через вторую - чистый газ-носитель
Рис. 23.3. Мост Уитстона для катарометра При использовании катарометра газом-носителем должен быть гелий или водород, обладающие большой теплоёмкостью. Этим достигается высокая чувствительность определения, так как разность между теплопроводностью газа-носителя и любого другого соединения всегда оказывается большой.
Пламенно-ионизационный детектор представляет собой металлическую камеру, в корпус которой снизу введена горелка (рис. 23.4). Для работы данного детектора необходимы водород, который смешивается с элюатом и сгорает при выходе из горелки, и воздух - для обеспечения горения водорода. В детекторе имеются два электрода. Одним из них является сама горелка, второй электрод расположен над ней. Пламя чистого водорода практически не содержит ионов, поэтому фоновое сопротивление пространства между электродами очень велико, а сила тока очень мала. Если в пламя из колонки попадает органическое вещество, то оно ионизируется. Поскольку в пламени появляются носители электрического заряда, сопротивление межэлектродного пространства резко уменьшается, а сила тока возрастает. Термоионный детектор внешне похож на пламенно-ионизационный. Он имеет кварцевую горелку, на конце которой находится таблетка из соли щелочного металла (например, CsBr). При нагревании эта соль испаряется и в газовой фазе устанавливается равновесие: CsBr + H+ › Cs+ + HBr При попадании в пламя соединения, содержащего в составе молекулы атомы фосфора и некоторые другие гетероатомы, скорость образования ионов резко увеличивается и сила тока возрастает. Термоионный детектор наиболее чувствителен к фосфорсодержащим соединениям. В меньшей степени он реагирует на соединения азота, серы, галогенов (кроме фтора), мышьяка, олова. Детектор электронного захвата представляет собой ионизационную камеру, в которой находится источник b-излучения, например, 63Ni или титановая фольга, содержащая адсорбированный тритий (рис. 23.5). В качестве газа-носителя при работе с детектором электронного захвата применяют азот, гелий, аргон и другие газы, способные ионизироваться с освобождением электрона. Фоновый ток детектора обусловлен, в основном, электронами. Молекулы анализируемых веществ, обладающие большим сродством к электрону, при попадании в детектор захватывают электроны и превращаются в анионы. Число носителей заряда при этом не изменяется, но сила тока уменьшается, так как анионы обладают на несколько порядков меньшей подвижностью, чем свободные электроны. Кроме того, образовавшиеся анионы вступают во взаимодействие с катионами газа-носителя, что вносит дополнительный вклад в уменьшение силы тока.
Рис. 23.5. Схема детектора электронного захвата Индексы удерживания Ковача Для идентификации веществ в хроматографии наряду с временем удерживания и удерживаемым объёмом используется параметр, называемый индексом удерживания. В газовой хроматографии для определения индекса удерживания в качестве стандартов берут два соседних н -алкана, один из которых элюируется до исследуемого соединения, а второй после (рис. 23.6).
Рис. 23.6. Определение индекса удерживания Ковача Логарифмический индекс удерживания равен:
где z - число атомов углерода в молекуле н -алкана, который элюируется первым Затем по справочным таблицам можно определить круг веществ, которые имеют близкую к рассчитанной величину индекса Ковача Практическое применение Газовую хроматографию используют для разделения, идентификации и количественного определения различных соединений, в том числе и лекарственных веществ, которые обладают достаточной летучестью (перегоняются без разложения в интервале температур до 400 °С). Методом ГХ можно определять и малолетучие вещества, если известен способ их переведения в летучие производные.
Газовая хроматография может быть использована для определения веществ, разрушающихся при нагревании, если процесс термического разрушения вещества хорошо воспроизводим. ГЛАВА 24
Общая характеристика Жидкостная хроматография - группа хроматографических методов, в которых подвижной фазой является жидкость.
В качестве сорбентов в жидкостной хроматографии применяют:
К неполярным относят также сорбенты с привитыми неполярными группами, например, химически модифицированные кремнезёмы с привитыми алкильными группами, содержащими от 2 до 18 углеродных атомов (рис. 24.1). В качестве подвижной фазы в жидкостной хроматографии используют воду, водные растворы различных веществ (сильные кислоты, кислотно-основные буферы и т.д.), органические растворители (спирты, ацетонитрил, тетрагидрофуран, диоксан, диэтиловый эфир, алканы и т.д.), а также водно-органические смеси.
Рис. 24.1. К ремнезём с привитыми октадецильными группами Плоскостная хроматография В плоскостной хроматографии подвижная фаза перемещается в плоском слое сорбента.
Как в БХ, так и в ТСХ разделение может быть обусловлено различными механизмами, например, адсорбционным, распределительным, ионообменным, ион-парным, адсорбционно-комплексообразовательным. Бумажная хроматография имеет ряд существенных недостатков и поэтому в настоящее время используется сравнительно редко: · процесс разделения зависит от состава и свойств бумаги; · содержание воды в порах бумаги может изменяться в зависимости от условий хранения; · очень низкая скорость хроматографирования (процесс получения хроматограммы может занимать нескольких суток), · низкая воспроизводимость результатов. В тонкослойной хроматографии обычно используют хроматографические пластины заводского изготовления с закреплённым слоем сорбента. Основа пластинки может быть изготовлена из алюминиевой фольги, полимера (например, полиэтиленгликольтерефталата), стекла. Для удерживания слоя сорбента на подложке применяется гипс, крахмал, силиказоль и др. Толщина слоя сорбента может быть различной (0,1 мм и более), но обязательно одинаковой в любом месте хроматографической пластинки. В качестве сорбентов в ТСХ используют силикагель, кизельгур, оксид алюминия, целлюлозу и др. В ионообменных хроматографических пластинках адсорбентами являются различные ионообменники (см. далее). В качестве подвижной фазы применяют либо индивидуальные растворители, либо смеси веществ, взятых в определённом соотношении.
24.2.1. Методика получения плоскостной хроматограммы Методика получения плоскостных хроматограмм включает в себя следующие этапы: · предварительный этап - подготовка сорбента и исследуемой пробы, подготовка подвижной фазы, насыщение хроматографической камеры; · нанесение исследуемой пробы на хроматографическую пластинку или бумагу; · хроматографирование; · высушивание хроматограммы; · обнаружение пятен (зон) разделённых компонентов пробы. Нанесение исследуемого раствора на хроматографическую пластинку или бумагу проводят градуированным капилляром, микрошприцом или микропипеткой. Капля наносится касанием капилляра или иглы поверхности пластинки (но не надавливанием, так как при этом можно повред
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-13; просмотров: 1904; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.15 (0.021 с.) |

 .
. ,
, ,
, - число волн, приходящихся на 1 см в вакууме.
- число волн, приходящихся на 1 см в вакууме.  , где l - длина волны (см). Размерность
, где l - длина волны (см). Размерность  - см-1.
- см-1.
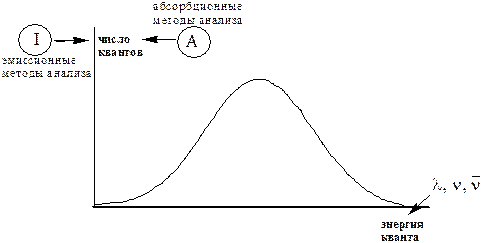




 или
или 





 можно разложить в ряд Маклорена
можно разложить в ряд Маклорена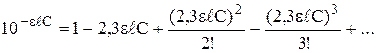
 (оптическая плотность раствора) невелико, то
(оптическая плотность раствора) невелико, то  и тогда
и тогда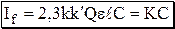




 Самый эффективный и в настоящее время практически единственный способ получения хроматограммы в количественном анализе
Самый эффективный и в настоящее время практически единственный способ получения хроматограммы в количественном анализе
 Используется, в основном, для разделения макроколичеств веществ в препаративных целях.
Используется, в основном, для разделения макроколичеств веществ в препаративных целях.
 Метод использовался на ранних стадиях развития хроматографии.
Метод использовался на ранних стадиях развития хроматографии.
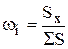 или
или  ,
где f – поправочные коэффициенты, учитывающие неодинаковую чувствительность детектора к различным компонентам смеси
,
где f – поправочные коэффициенты, учитывающие неодинаковую чувствительность детектора к различным компонентам смеси

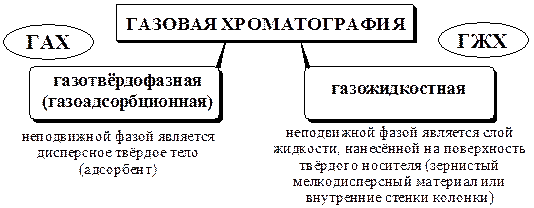

 Рис. 23.2. Схема катарометра
Рис. 23.2. Схема катарометра
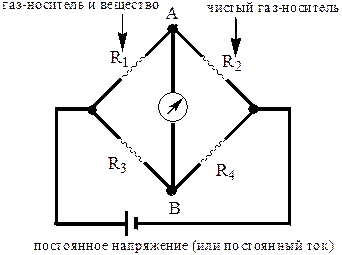
 Рис. 23.4. Схема пламенно-ионизационного детектора
1 - собирающий электрод; 2 - горелка
Рис. 23.4. Схема пламенно-ионизационного детектора
1 - собирающий электрод; 2 - горелка