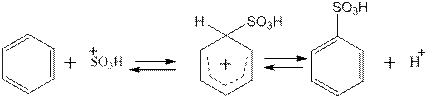Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Использование реакций электрофильного замещения.Содержание книги
Поиск на нашем сайте
Реакции электрофильного замещения условно обозначаются символом Se- моно записать эти реакции в виде уравнения: А:В + Х+ → A:X + B+ Здесь А:В обозначает молекулу субстрата, две точки — электронную пару связи А—В или А—X, Х+ — электрофильный реагент. Реакции электрофильного замещения наиболее характерны для соединений ароматического ряда. Важнейшие реакции электрофильного замещения в ароматическом ряду: C6H6 + HNO3 + H+ → C6H5NO2 + H2O Нитрование C6H6 + H2SO4 → C6H5SO3H + H2O Сульфирование
HOC6H6 + C6H5N2Cl → HOC6H4N=NC6H5 Азосочетание Склонность ароматических соединений к реакциям замещения объясняется тем, что этот тип взаимодействия, в отличие от реакций присоединения, не ведет к нарушению устойчивой ароматической системы и, следовательно, не требует добавочной энергии на преодоление энергии стабилизации этой системы. Реакции Se представляют собоймногостадийные процессы, механизм которых можно изобразить следующей схемой. Сначала под воздействием катализатора реагент X—Y переходит в активную форму Х+: X:Y + Катализатор → Х++ [Y: Катализатор]- Далее следует взаимодействие катиона Х+ с молекулой ароматического соединения, например, бензола:
Первая стадия, образование так называемого π - комплекса, включает взаимодействие между электрофильным реагентом Х+ и делокализованными π - электронами ядра. Эта стадия осуществляется быстро ивсегда обратимо. При этом происходит лишьнезначительное нарушение π - электронного облака бензольного кольца. Вторая стадия состоит в перестройке π - комплекса в карбениевый ион, так называемый π - комплекс, в котором реагент Х+ связан с определенным углеродным атомом ядра. В π - комплексе ароматический секстет нарушен: четыре π - электрона делокализованы в сфере воздействия пяти углеродных атомов. Шестой углеродный атом переходит при образовании π - комплекса из состояния sp2 в состояние sp3 гибридизации. Третья стадия реакции — отщепление протона и образование молекулы замещенного бензола. Отщеплению протона помогает основание В-, в роли которого может выступать растворитель или комплекс [Y-Катализатор]-. Обратимыми являются реакции сульфирования и алкилирования ароматических соединений. Если же в σ - комплексе образуются прочные π - связи между ядром и реагентом, обратный процесс, переход от σ-комплекса к π - комплексу, не наблюдается и реакция протекает необратимо. Это имеет место, в частности, при нитровании, галогенировании и ацилировании ароматических соединений. Скорость реакции электрофильного замещения водорода в ароматическом ядре меняется при переходе от бензола к его гомологам и производным: она зависит от числа и вида уже имеющихся в ядре заместителей. Реакция электрофильного замещения осуществляется тем легче, чем больше нуклеофильность ароматического ядра. Заместители электронодонорные, повышающие общую электронную плотность в ядре, облегчают реакцию электрофильного замещения и являются, таким образом, активирующими, а заместители электроноакцепторные, понижающие электронную плотность в кольце, этой реакции препятствуют, т. е. являются дезактивирующими. К электронодонорным (активирующим) заместителям относятся следующие атомы и группы атомов (в скобках указаны основные электронные эффекты, проявляемые этими заместителями): алкилы (+I), —ОН, —OR, —NH2, —NHR, — NR2, (+M, - I), — О-, (+М и +I). К электроноакцепторным (дезактивирующим) заместителям относятся: —COR, —СООН, -COOR, -CN, -NО2 (—М и -I), галогены (+М, - I), NR+ (—I). Пользуясь такой классификацией, можно заранее предсказать, что, например, толуол и фенол будут легче нитроваться и сульфироваться, чем бензол, а для введения в реакцию нитрования нитро- или бромбензола потребуются более жесткие условия, чем для аналогичной реакции с бензолом.
 
B случае электронодонорных заместителей с - I и +М - эффектами энергия активации образования σ - комплекса для орто- и пара замещения оказывается ниже, чем для мета замещения, а потому соответствующие продукты (орто- и пара изомеры) образуются скорее, чем мета изомеры. Электронодонорные заместители являются, таким образом, орто-, пара ориентантами. Электроноакцепторные заместители (обладающие —I- и - М-эффектами) дестабилизируют σ - комплекс, и это их влияниев наибольшей степени проявляется тогда, когда заместитель непосредственно связан с положительно заряженным углеродным атомом, поэтому:
 
Соответственно энергия активации для мета замещения оказывается несколько меньше, чем для орто- и пара замещения и соответствующий изомер образуется с большей скоростью. Электроноакцепторные заместители являются мета ориентантами. Заместители, обладающие - I- и +М-эффектами, в положительно заряженном и поэтому сильно электрофильном σ-комплексе проявляют главным образом свой +М-эффект. С его помощью они понижают энергию активации образования σ - комплекса с заместителями в орто- и пара положениях. Это справедливо также дляслучая заместителей типа галогенов. Таким образом, галогены, подобно электронодонорным заместителям, являются орто -, пара ориентантами. Поскольку они одновременно являются дезактивирующими заместителями, энергия активации образования σ -комплекса с галогенбензолами оказывается выше, чем с незамещенным бензолом, но ниже, чем соответствующая энергия σ -комплекса с соединениями, имеющими в ядре заместители с - I- и - М-эффектами. Если в ароматическом ядре имеется не один, а несколько заместителей, место вступления нового заместителя будет определяться суммарным влиянием уже имеющихся. При этом возможны два случая: влияние имеющихся заместителей может быть согласованным или несогласованным. Согласованное влияние оказывают заместители одного рода (орто-,, пара- или мета ориентирующие), когда они находятся в мета положении друг к другу, например:
Если в ядре находятся заместители разного рода (один орто, пара- а другой мета ориентирующий), их совместное влияние будет согласованным только при их нахождении в орто- или пара положении, например:
 
Несогласованная ориентация имеет место в тех случаях, когда заместители одного рода находятся в орто- или пара положениях по отношению друг к другу, или когда заместители разного рода оказываются в мета положении. В этом случае место вступления нового заместителя определяется относительной ориентирующей (активирующей) силой заместителей. Например, при нитровании о -метоксиацетанилида под влиянием ориентирующего действия амидной группы образуется в основном 2-метокси-4-нитроацетанилид:
Рассмотренные правила ориентации справедливы лишь для кинетически контролируемых процессов. В равновесных условиях преимущественно образуются наиболее стабильные изомеры (термодинамический контроль). Так, при алкилировании толуола хлористым алкилом в присутствии А1С13 при низкой температуре образуется в основном пара алкилтолуол, а при 60° С — более устойчивый мета изомер:
Лабораторная работа №14 «Сульфирование» Реакции, сопровождающиеся замещением водорода ядра сульфогруппой, относятся к сульфированию. Открытие реакции сульфирования принадлежит В. Бранду, который в 1819 г. осуществил синтез сульфопроизводного нафталина. Специфика этого процесса в отличие от других электрофильных реакций заключается в том, что он носит необратимый характер:
В зависимости от условий сульфирования в качестве сульфирующих агентов могут выступать различные частицы, однако во всех случаях процесс протекает через стадию образования σ-комплекса:
Наиболее распространенным сульфирующим агентом является серная кислота различной концентрации. Для трудно сульфируемых соединений применяют олеум с различным содержанием оксиды серы (VI). В этом случае первой стадией процесса является образование π - комплекса между π- электронной системой бензольного ядра (донор электрона) и электрон-дефицитным атомом серы в молекуле серного ангидрида SO3:
В ходе реакции π – комплекс перегруппировывается в σ-комплекс:
При дальнейшем течении процесса σ - комплекс с переносом протона от атома углерода к атому кислорода в экзотермической реакции превращается в бензолсульфокислоту:
Сульфирующим агентом служит также хлорсульфоновая кислота ClSO3H (монохлорангидрид серной кислоты). Для проведения сульфирования в газовой фазе используют триоксид серы. Сульфирование соединений, требующих мягких условий, осуществляют пиридинсульфотриоксидом C6H5N SO3.
Практическая часть
|
||||||||
|
|
Последнее изменение этой страницы: 2016-04-18; просмотров: 734; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.108 (0.007 с.) |

 Галогенирование
Галогенирование Алкилирование
Алкилирование Ацилирование
Ацилирование