
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Насичені галогенопохідні вуглеводнівСодержание книги
Поиск на нашем сайте ЗМІСТ
ГАЛОГЕНПОХІДНІ ВУГЛЕВОДНІВ
Галогенпохідні вуглеводнів – сполуки, в яких один або більше атомів Гідрогену заміщені на атом (чи атоми) галогену (галогенів).
Класифікація
1. Моногалогенопохідні:
2. Полігалогенопохідні:
НАСИЧЕНІ ГАЛОГЕНОПОХІДНІ ВУГЛЕВОДНІВ (ГАЛОГЕНАЛКАНИ) Ізомерія і номенклатура В залежності від структури атома Карбону, з яким зв’язаний галоген, розрізняють первинні, вторинні і третинні галогенопохідні. Їх ізомерія залежить від: а) ізомерії карбонового ланцюга; б) положення атома галогену в лацюзі. Номенклатура:
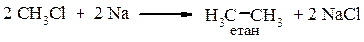
Способи одержання Галогенування алканів. (Активність галогенів знижується у ряду: F > Cl > Br > I).
2. Приєднання галогеноводнів до ненасичених сполук: а) за правилом Марковникова:
Механізм реакції АЕ – електрофільне приєднання. б) проти правила Марковникова (перексидний ефект Хараша):
3. Заміщення гідроксильної групи в спиртах при дії: а) тіонілхлориду:
б) п’ятихлористого фосфору:
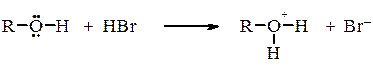
в) галогеноводнів:
4. Реакції обміну атомів галогену, які базуються на різній розчинності солей у певних розчинниках, наприклад:
Хімічні властивості
Галогенопохідні – один з найбільш реакційно здатних класів органічних сполук. Найбільш характерні для них реакції нуклеофільного заміщення (SN) та відщеплення (Е). ІІІ. Взаємодія з металами
ІV. Відновлення галогеналканів
НЕНАСИЧЕНІ ГАЛОГЕНОПОХІДНІ ВУГЛЕВОДНІВ Класифікація 1. Ненасичені галогенопохідні з атомом галогену, який сполучений з атомом Карбону безпосередньо біля подвійного зв’язку:
2. Ненасичені галогенопохідні з атомом галогену біля a-атома Карбону відносно подвійного зв’язку:
3. Ненасичені галогенопохідні з атомом галогену, який ізольований від подвійного зв’язку більше, ніж на один атом карбону:
У випадку (3) атом Хлору ізольований від подвійного зв’язку sp3-гібридними атомами Карбону, тому їх взаємний вплив не відбувається і такі сполуки повторюють хімічні властивості звичайних галогеналканів.
Хлористий вініл Одержання хлористого вінілу а) дегідрогалогенування дигалогенопохідних при дії спиртового розчину лугу:
б) термічне хлорування етилену:
в) приєднання хлористого водню до ацетилену:
Хлористий аліл 1. Одержання хлористого алілу – термічне хлорування алкенів в алільне положення:
Галогенування алкіларенів
Реакція хлорметилювання
Реакції сульфогрупи - утворення солей С6H5SO3H + NaOH ® C6H5SO3Na + H2O; - утворення хлорангідридів (сульфохлоридів) С6H5SO2OH + PCl5 ® C6H5SO2Cl + POCl3 + HCl. бензолсульфохлорид Проте, звичайно сульфохлориди одержують дією хлорсульфонової кислоти на вуглеводні: С6H5SO2OH + HOSO2Cl ® C6H5SO2Cl + H2SO4 Сульфохлориди можна легко перетворити в естери та аміди, подібно до хлорангідридів карбонових кислот: C6H5SO2Cl + NaOC2H5 ® C6H5SO2OC2H5 + NaCl етиловий естер бензолсульфокислоти C6H5SO2Cl + 2 NH3 ® C6H5SO2NH2 + NH4Cl Бензолсульфамід - утворення тіофенолів
ОДНО- І БАГАТОАТОМНІ СПИРТИ Спирти - це похідні вуглеводнів, у молекулах яких один або декілька атомів Гідрогену заміщені гідроксильними групами. В залежності від кількості гідроксильних груп спирти бувають одноатомні та багатоатомні.
Методи одержання спиртів Гідроліз галогенопохідних Спирти одержують гідролізом галогенпохідних при нагріванні з водою або водним роззином лугу. В першому випадку реакція зворотня
Металоорганічний синтез а) первинні спирти
б) вторинні спирти
в) третинні спирти
4. Каталітичне гідрування карбонільних сполук у присутності каталізаторів (Ni, Co, Pt, Pd) а) гідрування альдегідів і естерів з утворенням первинних спиртів:
б) каталітичне відновлення кетонів призводить до утворення вторинних спиртів:
в) відновлення ароматичних альдегідів і кетонів та естерів ароматичних кислот відбувається аналогічно. Окиснення алканів Реакція окиснення має практичне застосування для вищих парафінів С12-С20:
Будова спиртів
Зв’язки С–О і О–Н поляризовані, негативним кінцем диполя є Оксиген. Загальний (сумарний) дипольний момент спиртів m = 1,6-1,8 D. Полярність зв’язку О–Н значно вища, ніж полярність зв’язку С–О. Такий розподіл електронної густини сприяє тому, що для О–Н групи характерні гетеролітичні (іонні) реакції, в яких може розірватись О–Н-зв’зок або С–О. Полярність зв’язку О–Н і наявність неподіленої електронної пари електронів у атома Оксигену зумовлюють утворення водневих зв’язків, які сприяють існуванню асоціатів між молекулами спирту. Це пояснює високі температури кипіння спиртів (tкип.(С2Н5ОН) = 78,3 °С; tкип.(СН3–О–СН3) = 23,6 °С).
Спирти С1–С3 змішуються з водою у будь-яких попорціях, що пояснюється утворенням водневих зв’язків з молекулами води.
Хімічні властивості спиртів
1. Спирти – амфотерні сполуки. Виявляють властивості дуже слабких кислот: у реакції з металічним натрієм утворюють солі – алкоголяти:
Алкоголяти легко розкладаються водою, адже спирти більш слабкі кислоти ніж вода
Кислотні властивості спиртів зменшуються в наступному порядку: первинні > вторинні > третинні. Як і вода, спирти проявляють і основні властивості: реагують на холоду з сильними кислотами з утворенням солей алкілоксонію:
При нагріванні, а у випадку третинних спиртів на холоду, оксонієві солі перетворюються в галогенопохідні. Внутрішньо- та міжмолекулярна дегідратація спиртів Дегідратація При дії на спирти дегідратуючих речовин (H2SO4, H3PO4, Al2O3, CuSO4 та ін.) проходить міжмолекулярне або внутрішньомолекулярне відщеплення води з утворенням відповідно етерів та ненасичених сполук.
Внутрішньомолекулярна дегідратація проходить на тих самих каталізаторах, що і міжмолекулярна але при підвищеній температурі. В залежності від будови спирта і умов проведення реакції відщеплення води проходить за механізмами Е1 або Е2, наприклад для третинного спирту при каталітичній дії кислоти механізм реакції можна представити наступним чином:
3. Заміщення гідроксильної групи. Гідроксильна група спиртів здатна заміщуватися галогеном при взаємодії спиртів з галогеноводнями, галогенідами фосфору або тіонілхлоридом (див. розд. галогенопохідні вуглеводнів). Спирти порівняно легко окиснюються такими окисниками як KMnO4, K2CrO7. Первинні спирти окиснюються до альдегідів, вторинні до кетонів. Багатоатомні спирти повторюють властивості одноатомних.
ФЕНОЛИ. ХІНОНИ Фенолами називають органічні сполуки, які містять гідроксильну групу, безпосередньо сполучену з атомом Карбону бензольненового ядра. Залежно від кількості гідроксильних груп в їх структурі феноли поділяють на одноатомні, двоатомні, тощо. Методи одержання фенолів Гідроліз галогенопохідних
Реакцію проводят в присутності солей Купруму з 8%-ним розчином NaOH в автоклаві. Розклад гідропероксидів
4. Окиснювальне декарбоксилювання карбонових кислот. Реакція проходить при 200-300°С у присутності солей купруму (ІІ).
Розклад солей діазонію
Будова фенолів При розгляді будови фенолу можна було б припустити існування кето-єнольної таутомерії:
Однак більшість фенолів існує виключно в єнольній формі. Більша стійкість єнольній формі фенолів зумовлена більшим ступенем супряження неподіленої пари електронів атома Оксигену групи О–Н з p-електронами фенольного ядра, що призводить до стабільності молекули в порівнянні з кетонною формою. Хімічні властивості фенолів Хімічні властивості фенолів визначаються гідроксильною групою і сполученим з нею бензеновим ядром. Гідроксил виявляє –І-ефект і разом з тим вільні пари р-електронів атома Оксигену взаємодіють з π-електронами бензенового ядра (р,π-супряження), тому в результаті такої взаємодії р‑електронна густина Оксигену зміщується в напрямку бензенового ядра (ефект супряження сильніший ніж індукційний).
1. Кислотні властивості фенолів. Феноли виявляють більшу кислотність ніж спирти та вода, проте вони слабкіші ніж вугільна та карбонові кислоти. Феноли реагують з розчинами лугів з утворенням фенолятів. В свою чергу феноли виділяють з розчинів фенолятів вугільною кислотою: ArOH + NaOH ® ArONa + H2O ArONa + CO2 + H2O ® ArOH + NaHCO3 Більша кислотність фенолів у порівнянні зі спиртами пояснюється додатковою делокалізацією заряду у фенолят-аніоні, якої немає в алкоголят аніоні:
На кислотні властивості фенолу впливають замісники в ароматичному ядрі. Електроноакцепторні замісники особливо в орто - і пара - положеннях, значно збільшують кислотність фенолів. Наприклад, тринітрофенол (пікринова кислота) має константу іонізації 1,6·10-1. 2. Утворення етерів та естерів. Феноли легко алкілуються при діі на феноляти галогенопохідних, особливо у присутності порошку Купруму, а також при дії алкілсульфатів, естерів сульфокислот та діазометану: С6H5ONa + C2H5I ® С6H5OC2H5 + NaI фенетол С6H5OH + CH2N2 ® С6H5OCH3 + N2 анізол При взаємодії фенолятів з галогенаренами утворюються ароматичні етери: С6H5ONa + С6H5Br ® С6H5OC6H5 + NaBr дифеніловий етер Дифеніловий етер має запах герані. Взаємодія фенолятів з галогенагідридами карбонових кислот призводить до утворення естерів: С6H5ONa + CH3COCl ® С6H5OOCH3 + NaCl Аналогічні продукти одержуються також за реакцією ацилування фенолів ангідридами карбонових кислот. Можлива естерифікація фенолу кислотами в присутності H2SO4 та H3PO4 в умовах азеотропної відгонки води. 3. Окиснення фенолів. Феноли легко окиснюються, навіть киснем повітря. При окисненні хромовою сумішшю фенол перетворюється в п ‑бензохінон, який забарвлений у жовтий колір.
Багатоатомні феноли Три ізомерні двоатомні феноли
а також три ізомери триатомних фенолів:
мають властивості близькі до властивостей одноатомних фенолів: утворюють етери та інші похідні за гідроксильною групою, дають кольорові реакції з FeCl3, але легше окиснюються до відповідних хінонів і є більш сильними кислотами. Резорцин і флороглюцин мають здатність до кето-єнольної таутомерії:
Хінони Хінони – циклічні a,b-ненасичені дикетони. Найпростішими представниками є о - і п -хінони:
Хінони –легко утворюються з двоатомних фенолів і легко відновлюються до двоатомних фенолів. Хінони вступають у реакції характерні для кетонів. Крім того хінони вступають у реакції 1,4- і 1,6-приєднання, які призводять до утворення похідних двоатомних фенолів. При змішуванні рівномолярних кількостей хінону і гідрохінону утворюється молекулярна сполука – хінгідрон.
АЛЬДЕГІДИ І КЕТОНИ Альдегіди і кетони – це сполуки, у молекулах яких міститься функціональна група >C=O (карбонільна або оксогрупа). В альдегідах карбонільна група сполучена з карборадикалом і одним атомом Гідрогену, у кетонах вона сполучена з двома карборадикалами: Класифікація
Способи одержання Реaкції окиснення - ненасичених вуглеводнів
- насичених вуглеводнів
- ароматичних вуглеводнів
- спиртів
Хімічні властивості 1. Особливості реакцій нуклеофільного приєднання до альдегідів і кетонів а) Електронні фактори пов'язані з тим, що алкільна група, як донор електронів подає електронну густину на атомі Карбону карбонільної групи, що знижує частково позитивний заряд (δ +) на ньому.
У випадку альдегідів таке зниження δ + проходить менше, так як тільки одна алкільна група. Для кетону таке зниження δ + на карбонільному атомі Карбону проходить більше, так як в цьому випадку має місце вплив двох алкільних груп. б) Просторові фактори. При приєднанні нуклеофілу до карбонільної групи атом Карбону переходить з sp2–гібридного стану до sp3–гібридного стану. Валентні кути змінюються від 120° до 109°28'. Тобто проходить зближення замісників зв'язаних з карбонільною групою. Для альдегідів таке зближення проходить легко, так як один із замісників – це атом Гідрогену. Для кетонів таке зближення проходить трудніше і при наявності особливо об'ємних замісників (наприклад, трет -бутильних) реакційна здатність кетонів знижується настільки, що реакції нуклеофільного приєднання стають неможливими. Тому альдегіди є більш реакційноздатними сполуками ніж кетони. Здатність альдегідів і кетонів вступати в реакції приєднання, приєднання-відщепленні визначається величиною частково позитивного заряду на атомі карбону карбонільної групи – чим більше δ+ на атомі Карбону тим більшу реакційну здатність мають альдегиди та кетони у вище зазначених реакціях. Тому, реакційна здатність карбонільних сполук зменшується в такому ряду:
В цілому: а) альдегіди вступають в реакцію легше кетонів; б) аліфатичні сполуки вступають в реакції легше алкілароматичних і тим більше чисто ароматичних. Ароматичні альдегіди і кетони, які містять в о - і особливо в п -положеннях до карбонільної групи електоноакцепторні замісники мають більш високу реакційну здатність за рахунок збільшення частково позитивного заряду (δ+ ) на атомі Карбону карбонільної групи і навпаки: введення електронодонорних замісників в тіж положення зменшують реакційну здатність альдегідів і кетонів за рахунок зниження заряду (δ+)на атомі Карбону карбонільної групи. Головна відзнака альдегідів від кетонів – альдегіди окиснюються дуже легко, особливо ароматичні, а кетони – важко. Для альдегідів і кетонів характерні реакції нуклеофільного приєднання води, спиртів, синильної кислоти, гідросульфіту натрію, реактиву Гріньяра; реакції з азотовмісними сполуками (амоніаком, гідроксиламіном, гідразином тощо); реакції конденсації (альдольна, кротонова), окиснення. Реакції полімеризації Реакції полімеризації характерні лише для альдегідів, в першу чергу для формальдегіду. а) циклічна полімеризація формальдегіду у присутності кислот:
б) в присутності каталізатора на зразок оксиду алюмінію або карбоніла феруму в органічних розчинниках проходить лінійна полімеризація:
Поліформальдегід за звичайних умов не розчиняється в жодному розчиннику, має високу міцність. Застосовується для виробництва синтетичного волокна. в) здатність до циклічної полімеризації проявляє й оцтовий альдегід:
НІТРОСПОЛУКИ Похідні вуглеводнів в молекулах яких є група –NO2. Гомологічний ряд загальної формули R–NO2. Класифікація
Ізомерія і номенклатура В залежності від характеру атома Карбону з яким зв’язана нітрогрупа аліфатичні нітросполуки бувають:
Для нітросполук характерна структурна ізомерія, що обумовлена розгалуженням вуглецевого ланцюга та ізомерія положення нітрогрупи. Способи одержання нітросполук
Будова нітрогрупи
Нітрогрупа є одним із найбільш сильних акцепторів електронів, так як на атомі Нітрогену локалізований цілий позитивний заряд, що з одного боку призводить до різкого збільшення рухливості протону на α-атомі Карбону (більш сильному ніж цей ефект у альдегідах і кетонах), а з іншого боку до дезактивації ароматичного кільця до реакцій електрофільного заміщення і полегшення реакцій нуклеофільного заміщення. Хімічні властивості
І. Реакції за нітрогрупою Класифікація
Способи одержання амінів
Алкілування a. Алкілування амоніаку галоїдними алкілами
b. Алкілування аміаку спиртами
Реакції а) і b) призводять до утворення суміші первинних, вторинних і третинних амінів, а також до четвертинних амонійних солей. Реакція Гофмана
Відновлення ізоціанідів
За цією реакцією одержують вторинні аміни, які обов’язково містять метильну групу з одного боку. 2. Метод допоміжного ацилювання (для жирно-ароматичних амінів)
Алкілування амінів a. Галоїдними алкілами
b. Спиртами
Реакції а,b також приводять до утворення суміші вторинних і третинних амінів.
Арилювання вторинних амінів
Будова триметиламіну
Аліфатичні аміни аналогічно амоніаку мають пірамідальну будову, атом Нітрогену розташований у вершині піраміди. Кути між зв’язками становлять»106-108°. Атом Нітрогену у триметиламіні знаходиться в sp3-гібридному стані. Основність амінів Основні властивості амінів визначається наявністю на атомі Нітрогену пари електронів і її здатністю приєднувати протон. Аміни як правило є більш сильними основами ніж вода, так як Нітроген менш електронегативний елемент ніж кисень і відповідно легше віддає свою пару електронів для протонування. Основність амінів в значній мірі залежить від їх будови. Аліфатичні аміни є більш сильними основами ніж ароматичні. У аліфатичному ряді в протонних розчинниках вторинні аміни більш сильні основи ніж первинні, а третинні аміни мають менші основні властивості ніж вторинні, що пояснюється гіршою сольватацією розчинників через просторові фактори.
1. Водневі розчини алкіламінів мають лужне середовище:
2. З кислотами алкіламіни утворюють соли алкіламмонію:
3. Реакція алкілування – взаємодія з галогеналканами
4. Реакція ацилування – взаємодія амінів з ацилюючими реагентами Первинні та вторинні алкіламіни вступають в реакцію з функціональними похідними карбонових кислот (галогенангідридами, ангідридами, естерами) з утворенням N-заміщених амідів карбонових кислот. В процесі реакції атом Гідрогену біля Нітрогену в молекулі амінів заміщується на залишок карбонової кислоти. Третинні аміни не містять біля атомі Нітрогену атома Гідрогену и тому в цю реакцію не вступають.
5. Взаємодія з нітритною кислотою (реакція використовується для ідентифікації аліфатичних амінів) Первинні алкіламіни під дією нітритної килоти перетворюються у спирти з виділенням азоту та води:
Вторинні алкіл аміни в реакції з нитритною кислотою утворюють
Третинні аміни за звичаної температури з нітритною кислотою не реагують. 6. Ізонітрильна реакція. Специфічна реакція знаходження первинних амінів. При нагріванні первинних алкіламінів з хлороформом у присутності лугів у спиртовому середовищі утворюються ізонітрили (ізоціаніди):
Ізонітрили мають дуже неприємний запах. Ця реакція дуже чуттєва. Реакція окиснення Первинні алкіламіни при окисненні утворюють нітроалкани:
Під дією сильних окиснювачів первинні аліфатичні аміни утворюють суміш речовин, серед яких переважають альдегіди, а вторинні – тетразаміщені гідразини. Третинні аліфатичні аміни окиснюються атомарним киснем або пероксидом водню:
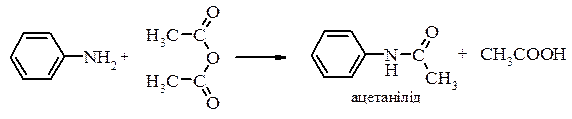
Реація ацилювання
Аніліди легко гідролізуються в кислому або лужному середовищах з утвренням вихідного аміну і карбонової кислоти:
Здатність ацильних похідних легко піддаватися гідролізу дозволяє використовувати цю реакцію в органічному синтезі для тимчасового захисту аміногрупи від окиснення та для зменшення її донорних властивостей. 3. Реакції утворення ізоціанідів (якісна реакція). Первинні ароматичні аміни при нагріванні з хлороформом та лугами в спиртовому середовищі утворюють ізоціаніди - речовини з неприємним запахом:
4. Реакції взаємодії з нітритною кислотою. Аліфатичні і ароматичні аміни по-різному реагують з нітратною кислотою. Цю реакцію можливо використовувати щоб розрізняти первинні, вторинні та третинні аміни алифітичного і ароматичного рядів. Первинні ароматичні аміни з нітритною кислотою в присутності сильної мінеральною кислоти утворюють солі діазонію. Ця реакція отримала назву реакції діазотування:
Вторинні ариламіни та N-алкілариламіни в цих умовах реагують аналогічно аліфатичним амінам і перетворюються у N-нітрозоаміни:
Третинні ароматичні аміни утворюють з нітритною кислотою ароматичні нітрозосполуки ─ відбувається реакція електрофільного заміщення за бензольним кільцем:
5. Реакція взаємодії з ароматичними альдегідами. Первинні ароматичні аміни реагують при нагріванні з ароматичними альдегідами, утворюя азометини (основи Шифа):
6. Реакції за участю атомів Карбону ароматичного ядра. За рахунок супряження електронів Нітрогену з ароматичним ядром значно збільшується електронна густина в орто - і пара -положеннях і утворюються більш стійкі σ-комплекси, що значно підвищують активність ароматичних амінів до реакції електрофільного заміщення в орто- і пара- положеннях. - Реакції галогенування. - Анілін взаємодіє з бромною водою при відсутності каталізаторів і утворюється 2,4,6-триброманілін.
- Реакція нітрування. У зв’язку з легким окисненням ароматичних амінів за аміногрупою проведення реакцій з реагентами, які можуть призвести до окиснення, проводять в умовах захисту аміногрупи реакцією ацилювання. Після проведення реакції ацильну групу гідролізують. Таким чином проводять реакцію нітрування.
- Реакція сульфування. При нагріванні аніліну з концентрованою сульфатною кислотою утворюється п ‑амінобензолсульфокислота, яку найчастіше назівають сульфаніловою кислотою. Реакція проходить через стадію утворення
7. Окиснення ароматичних амінів:
Реакції без виділення азоту
Реакції азосполучення, які призводять до утворення барвників. Азосполучення – це взаємодія солей арилдіазонію з похідними ароматичних вуглеводнів (амінів, фенолів). Азосполучення призводить до утворення азосполук, в молекулах яких присутня азогрупа –N=N–, яка знаходиться в супряженні з ароматичною π-електронною системою. Оскільки діазокатіон достатньо слабкий електрофіл, в реакцію азосполучення вступають вуглеводні, що мають сильні електронодонорі групи, наприклад, –NH2, –OH. Середовище реакційної маси при цьому не повинно бути дуже кислим, так як аміногрупа
Механізм:
Важлива властивість солей діазонію – це їх здатність вступати в реакції відновлення. Ця властивість солей діазонію використовується при встановленні будови азобарвника. Так при дії лужного розчину гідросульфіту натрію азобарвник легко розщеплюється на два аміни, що дає можливість встановити будову вихідного барвника.
КАРБОНОВІ КИСЛОТИ Карбонові кислоти – вуглеводні, які в своєму складі мають карбоксильну групу COOH. Загальна формула R–COOH За числом карбоксилів – одно- та багатоосновні. За природою вуглеводневого радикалу R бувають – насичені, ненасичені, аліфатичні та циклічні, ароматичні.
Окислення вуглеводнів
Окислення кетонів
Гідроліз галогенопохідних
Кислотні властивості
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-29; просмотров: 1442; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.214 (0.016 с.) |

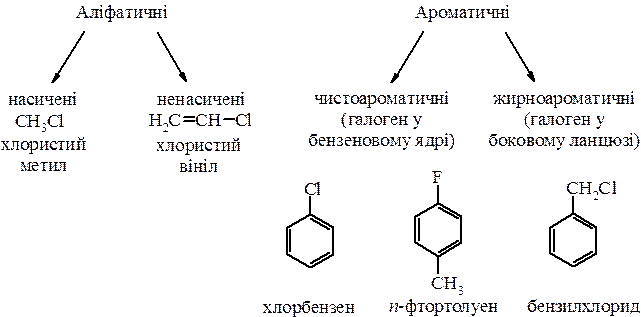













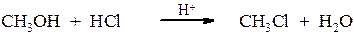
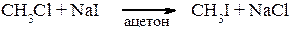
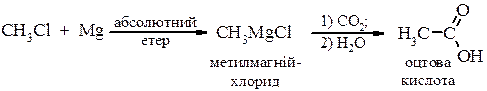





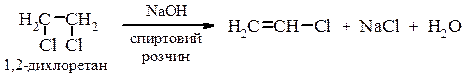



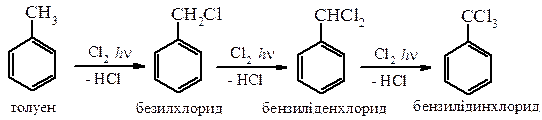

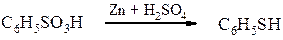
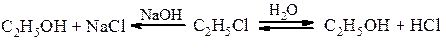
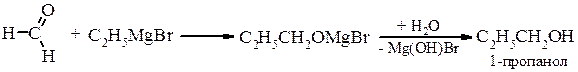
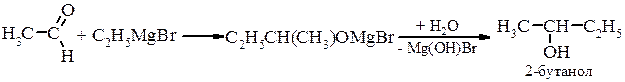
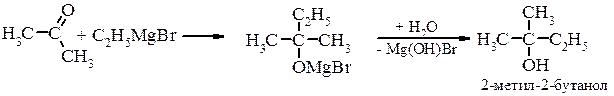
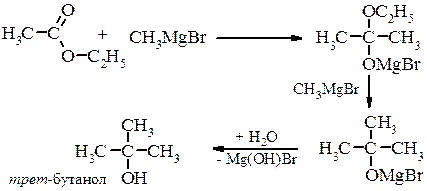
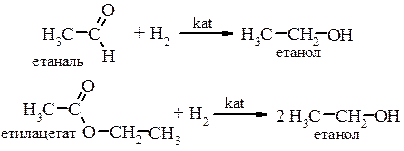

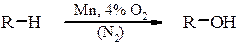
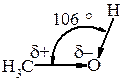
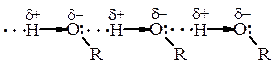


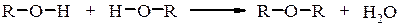
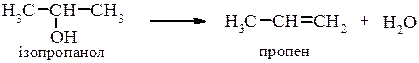
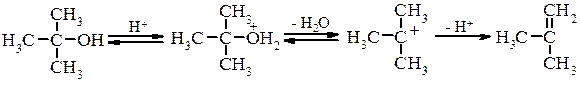


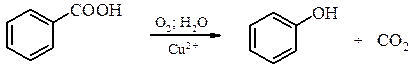

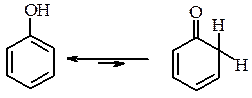
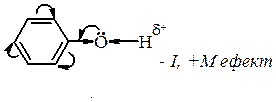


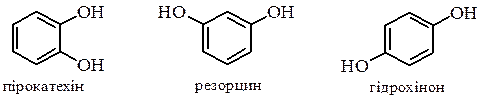
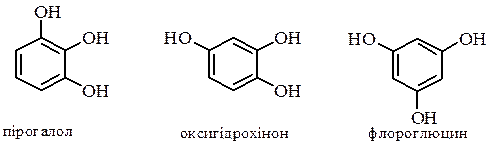 ,
,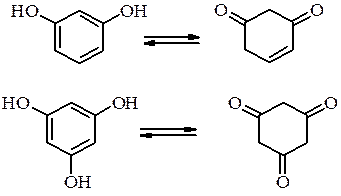








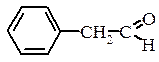





 2. Каталiтичне дегідрування спиртів
2. Каталiтичне дегідрування спиртів 3. Гідроліз гемінальних дигалогеналканів
3. Гідроліз гемінальних дигалогеналканів









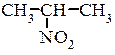
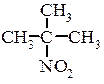

 метиламін
метиламін
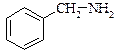 бензиламін
бензиламін
 анілін
анілін
 диметиламін
диметиламін
 N-метиланілін
N-метиланілін
 дифеніламін
дифеніламін
 триметиламін
триметиламін
 N,N-диметиланілін
N,N-диметиланілін
 трифеніламін
трифеніламін
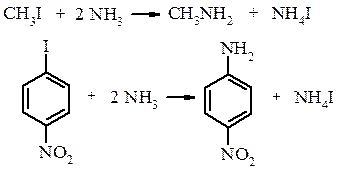
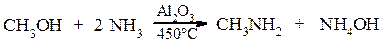
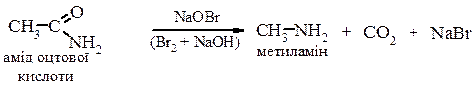

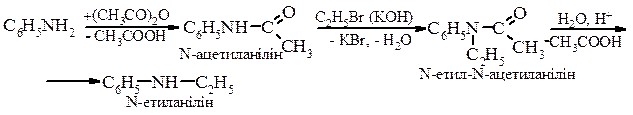


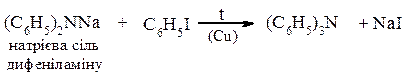












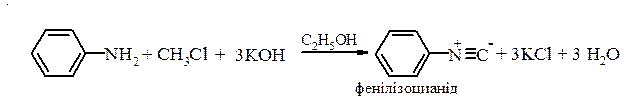
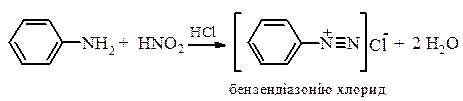
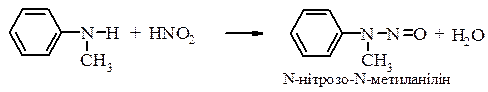
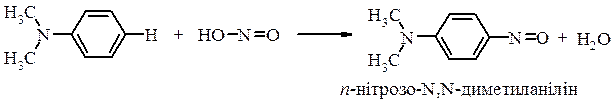
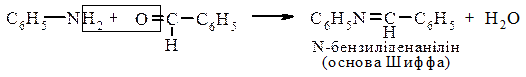
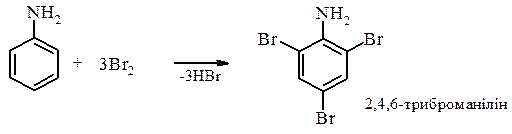
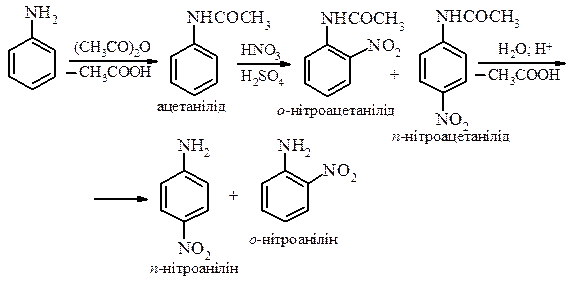
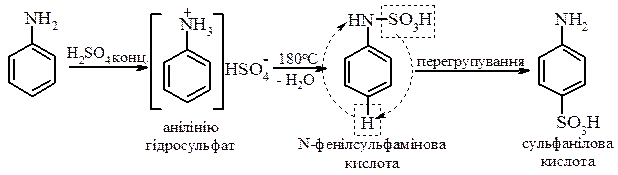
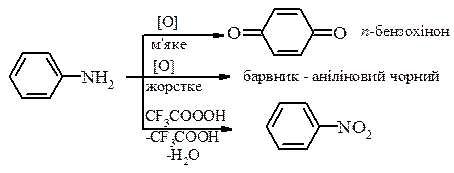

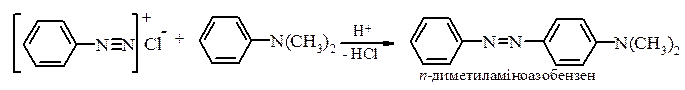
 Азобарвники застосовують для забарвлення самих різних матеріалів, в першу чергу, волокон (рослинних, тваринних та штучних) і тканин; також пластмас, гуми, деревини, шкіри і харчових продуктів. Вони використовуються для виготовлення лаків, покривних і поліграфічних барвників та як індикатори.
Азобарвники застосовують для забарвлення самих різних матеріалів, в першу чергу, волокон (рослинних, тваринних та штучних) і тканин; також пластмас, гуми, деревини, шкіри і харчових продуктів. Вони використовуються для виготовлення лаків, покривних і поліграфічних барвників та як індикатори.






