
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
По физико-химическим методам анализаСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте
МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ПРАКТИКУМУ ПО ФИЗИКО-ХИМИЧЕСКИМ МЕТОДАМ АНАЛИЗА ДЛЯ СТУДЕНТОВ 2 КУРСА СПЕЦИАЛЬНОСТИ «ХИМИЯ» С О Д Е Р Ж А Н И Е СПЕКТРОСКОПИЧЕСКИЕ МЕТOДЫ АНАЛИЗА МОЛЕКУЛЯРНАЯ СПЕКТРОСКОПИЯ 1.1.1. Порядок работы на фотоэлектроколориметре КФК-2. 1.1.2. Работа 1. Изучение колориметрической реакции. 1.1.3. Работа 2. Определение никеля (II) с диметилглиоксимом в присутствии окислителя методом градировочного графика. 1.1.4. Работа 3. Определение железа (III) c cульфосалициловой кислотой методом добавок. 1.1.5. Работа 4. Определение меди (II) дифференциальным методом. АТОМНАЯ СПЕКТРОСКОПИЯ 1.2.1. Работа 5. Определение железа в водопроводной воде. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА ПОТЕНЦИОМЕТРИЯ 2.1.1. Порядок работы на приборах 2.1.2. Работа 6. Определение соляной кислоты 2.1.3. Определение смеси соляной и уксусной кислот 2.1.4. Определение смеси бромид– и иодид–ионов 2.1.5. Определение железа (II). Электрогравиметрия 2.2.1. Определение меди (II) методом внутреннего электролиза. Экстракция 2.3.1. Экстракционные реакции Хроматография 2.4.1. Определение физических свойств ионита. 2.4.2. Определение обменной емкости ионита. 2.4.3. Определение общей концентрации электролита. 2.4.4. Определение меди и цинка при совместном присутствии. Молекулярная спектроскопия Фотометрический метод количественного определения основывается на способности определяемого вещества в оптически активной форме селективно поглощать электромагнитное излучение видимого участка спектра (400-780 нм). Общая схема фотометрического определения заключается в следующих стадиях: 1. Подготовка пробы и переведение вещества в раствор. 2. Получение окрашенной аналитической формы определяемого вещества в результате проведения реакции при оптимальных условиях, обеспечивающих максимальную избирательность и чувствительность. 3. Измерение светопоглощения раствора аналитической формы (регистрация аналитического сигнала). 4. Проверка результата анализа, оценка его воспроизводимости и определение окончательного результата со статистической обработкой. Порядок работы на фотоэлектроколориметре КФК-2. Фотоэлектроколориметр КФК-2 предназначен для измерения коэффициентов пропускания (Т,%) и оптической плотности (А) жидких растворов в отдельных участках спектрального диапазона длин волн 315-980 нм, выделяемых светофильтрами.
Принципиальная оптическая схема прибора.
1- осветитель 4,5- объектив 8- защитные стекла 2- конденсор 6- светофильтр 10- пластина-делитель 3- диафрагма 7- кювета 11- фотоприемники Характеристика светофильтров
Порядок работы. Подготовка к работе. 1) Колориметр включить в сеть (вилка и тумблер «сеть»). Открыть кюветное отделение. Прогрев прибора до начала измерений составляет 15 мин. 2) Ввести цветной светофильтр, рекомендуемый методикой, установив рабочую длину волны. 3) Установить минимальную чувствительность: а) ручку «чувствительность» - в положение «1»; б) ручку «установка 100 грубо» - в крайнее левое положение. Внимание! После завершения работ до выключения ручку «чувствительность» установить в положение «1» (красн.), а «установка 100 грубо» - в крайнее левое положение. После этого выключить тумблер «сеть». Измерение. 1) В кюветодержатель помещают кюветы с раствором сравнения (раст-воритель, «холостой раствор» или контрольный раствор, по отношению к которому ведутся измерения) и исследуемый раствор. 2) В световой поток поместить кювету с раствором сравнения. Закрыть кюветное отделение. 3) Ручками «чувствительность», «установка 100 грубо» и «установка 100 точно» выставить 100% пропускание по верхней шкале. Внимание! При измерении со светофильтрами, отмеченными на лицевой панели черным цветом, ручку «чувствительность» установить в положение 1-3, отмеченных также черным цветом, аналогично поступают и в случае светофильтров, отмеченных красным цветом. 4) Поворотом ручки кюветного отделения в световой пучок поместить кювету с исследуемым раствором. 5) Отклонение стрелки от нулевой отметки показывает значение: по верхней шкале – коэффициента пропускания Т, %; по нижней шкале – оптической плотности А.
Работа 1. Исследование колориметрической реакции. Условия проведения фотометрической реакции, приводящей к образованию соединения, поглощающего световую энергию в УФ или видимой области спектра, должны обеспечивать полноту образования светопоглощающего соединения и выполнение основного закона светопоглощения в широком диапазоне концентраций. Оптимизация условий фотометрических измерений предполагает: - выбор длины волны максимального светопоглощения. Для выбора длины волны λmax, которая соответствует наибольшей чувствительности данной фотометрической реакции необходимо получить спектральную характеристику изучаемого раствора, называемую спектром поглощения. - выбор толщины кюветы. - нахождение области линейности градуировочного графика как области подчинения фотометрической системы основному закону светопоглощения. Приборы и реактивы: Фотоэлектроколориметр КФК-2 хромат калия, 5%-ный раствор; рабочий раствор соли никеля(II), содержащий 0,01 мг/мл; йод, 0,05 М раствор; диметилглиоксим, 1%-ный раствор в 20%-ном растворе щелочи; HCl, 1М раствор; рабочий раствор соли меди, содержащий 1 мг/мл Cu2+; для приготовления рабочего раствора навеску 3,931 г CuSO4 ∙ 5H2O растворяют в 25 мл 2м раствора H2SO4, доводят объем раствора до 1,00 л дистиллированной водой; аммиак, 10%-ный раствор. Приготовление фотометрируемых растворов. 1) в мерные колбы, емкостью 50,0 мл помещают 1,0;2,0;3,0;4,0;5,0 мл 5%-ного раствора хромата калия и доводят объем до метки дистиллированной водой; 2) в мерные колбы, емкостью 50,0 мл помещают 2,0;4,0;6,0;8,0;10,0 мл стандартного раствора никеля, а также по 0,5мл раствора йода и по 0,5мл раствора диметилглиоксима и доводят дистиллированной водой до метки. 3) В мерные колбы емкостью 50,0мл помещают 1,0;2,0;3,0;4,0;5,0мл раствора меди, по 10,0мл раствора аммиака и доводят до метки дистиллированной водой. Определение рабочей длины волны. Для выбора длины волны, соответствующей максимальному светопоглощению, измеряют на фотоэлектроколориметре оптические плотности третьих растворов со всеми светофильтрами, кроме первого в кювете с ℓ=1см. Результаты записывают по следующей форме: Таблица 1. Выбор светофильтра для раствора хромата калия (СCrO42-=…мг/мл, ℓ=1см).
Таблица 2. Выбор светофильтра для раствора диметидглиоксимата никеля (СNi=…мг/мл, ℓ=1см).
Таблица 3. Выбор светофильтра для раствора аммиакатамеди (СCu=…мг/мл, ℓ=1см).
По данным таблиц 1-3 строят графики зависимости А=f(λ) и определяют λопт., для которой наблюдается максимальное светопоглощение. Определение интервала линейности градуировочного графика. Измеряют оптические плотности стандартных растворов на выбранной длине волны. Результаты измерений записывают по форме: Таблица 4. Оптические плотности растворов хромата калия (λопт.= … нм, ℓ=1см).
Таблица 5. Оптические плотности растворов диметилглиоксимата никеля (λопт.= … нм, ℓ=1см).
Таблица 6. Оптические плотности растворов аммиаката меди (λопт.= … нм, ℓ=1см).
По данным таблиц 4-6 строят градуировочные графики в координатах А=f(C, мг/мл) и определяют концентрационные области линейности. Рассчитывают чувствительность фотометрического определения как тангенс угла наклона градуировочного графика: H=ΔA/ΔС. Рассчитывают значение коэффициента светопоглощения для фотометрированных растворов по формуле: k =А/Сст.· ℓ Результаты расчета представляют по форме: Таблица 7. Коэффициент светопоглощения растворовхромата калия (λопт.= … нм, ℓ=1см).
Таблица 8. Коэффициент светопоглощения растворов диметилглиоксимата никеля (λопт.= … нм, ℓ=1см).
Таблица 9. Коэффициент светопоглощения растворов аммиаката меди (λопт.= … нм, ℓ=1см).
Значения коэффициентов чувствительности и средние значения коэффициентов поглощения для стандартных растворов: HCrO42- =…, kсредн.= …; HNi- =…, kсредн.= …; HCu=…, kсредн.= ….
Работа 2. Определение никеля с диметилглиоксимом в присутствии окислителей. Метод основан на измерении интенсивности красно-коричневой окраски растворов, которые образуются при взаимодействии ионов никеля (II) с диметилглиоксимом (DH) в присутствии окислителя. В качестве окислителей могут применяться йод, бром, персульфат аммония, пероксид водорода. В условиях проведения реакции происходит окисление никеля, вероятно, до состояния Ni3+. Образование комплекса наблюдается при молярном соотношении количеств Ni:DH = 1:3. Максимальное поглощение наблюдается при λ=470 нм, при этом ε=1.3∙104. Определению мешают вещества, имеющие собственную окраску и катионы, образующие осадки гидроксидов в щелочной среде (Fe,Al,Mg,Cu,Mn). Определение можно проводить в присутствии кобальта, ванадия, молибдена. Приборы и реактивы: стандартный раствор соли никеля(II), содержащий 0.1 мг/мл Ni(II); раствор соли никеля, содержащий 0.01 мг/мл (готовят разбавлением стандартного раствора в день употребления); йод, 0.05 М раствор; диметилглиоксим, 1%-ный раствор в 20%-ном растворе щелочи; HCl, 1М раствор; фотоэлектроколориметр КФК-2. Выполнение работы: Для приготовления эталонных растворов берут 5 мерных колб емкостью 50.0 мл, вводят в каждую 20.0 мл воды, стандартный раствор соли никеля, с содержанием никеля в мг: 0.02; 0.04; 0.06; 0.08; 0.10 соответственно, добавляют 2-3 капли HCl и растворы: 0.5 мл йода, 0.5 мл диметилглиоксима, объем доводят водой до метки колбы. Через 10 минут растворы фотометрируют и строят градуировочный график в координатах А=f(СNi). В качестве раствора сравнения берут воду. Результаты фотометрирования стандартных растворов записывают в таблицу: Таблица 1. Данные для построения градуировочного графика (λопт.=490 нм, ℓ=1см).
Для определения никеля испытуемый раствор помещают в мерную колбу емкостью 50.0 мл. Проводят те же операции с добавлением тех же количеств реагентов, как и при приготовлении стандартных растворов. Определение массы никеля в объеме колбы проводят интерполяцией по ранее построенному графику.
Работа 3. Определение железа (III) с сульфосалициловой кислотой в виде трисульфосалицилата методом добавок.
Железо (III) образует с сульфосалициловой кислотой ряд комплексных соединений в зависимости от кислотности раствора: при рН 1.8-2.5 образуется комплексное соединение фиолетового цвета, имеющее состав 1:1. Максимум светопоглощения моносульфосалицилата железа наблюдается при λ=510 нм молярный коэффициент светопоглощения ε510 =1.8∙103; при увеличении рН до 4-8 образуется комплексное соединение, имеющее состав 1:2; при рН 9.0-11.5 образуется комплекс состава 1:3, растворы которого окрашены в желтый цвет. Трисульфосалицилат железа имеет λmax=416 нм и ε416=5.8∙103; при рН >12 происходит разложение комплексного соединения и выпадение в осадок гидроксида железа. Железо (II) не дает с сульфосалициловой кислотой интенсивной окраски, но вследствие легкой окисляемости ионов железа (II) в железо (Ш) в щелочной среде, можно определять сумму железа (II) и железа (Ш). Комплексные соединения железа с сульфосалициловой кислотой более устойчивы, чем роданидные комплексы железа, что позволяет применять рассматриваемый метод для определения железа в присутствии фосфатов, ацетатов и боратов. В присутствии магния, алюминия, марганца и некоторых других элементов более применим способ определения железа в кислой среде.
Приборы и реактивы: стандартный раствор соли железа (III), содержащий 0.1 мг/мл Fe3+; сульфосалициловая кислота, 10%-ный раствор; серная кислота, 2н раствор; аммиак,10%-ный раствор; азотная кислота (1:1)
Расчетный вариант метода добавок. Выполнение работы. Берут два раствора в мерных колбах на 50.0 мл неизвестной концетрацией железа. К одному из них добавляют 50.0-100.0 мкг железа, в каждую колбу приливают 5.0 мл 10%-ного раствора сульфосалициловой кислоты, 5.0 мл 10%-ного раствора аммиака и доводят объем водой до метки колбы. Через 10 минут измеряют оптическую плотность каждого раствора относительно воды при λ=400 нм. Искомую концентрацию железа(III) вычисляют по формуле: Сx= где Сх – искомая концентрация железа (Ш), Ах- оптическая плотность исследуемого раствора; Ах+д- оптическая плотность исследуемого раствора с добавкой, Сд- концентрация добавки в исследуемом растворе, которую вычисляют по формуле: Сд= Графический вариант метода добавок. Берут 4 аликвоты исследуемого раствора в мерных колбах на 50.0 мл. В аликвоты 2,3 и 4 вводят известные, возрастающие добавки железа (Ш) в количестве 50.0; 100.0 и 150 мкг железа (Ш). Добавляют к каждому раствору по 5.0 мл 10%-ного раствора сульфосалициловой кислоты, затем по 5.0 мл 10%-ного раствора аммиака, доводят водой объем до метки колбы. Через 10 минут измеряют оптическую плотность всех растворов относительно воды и строят график в координатах А = f(Сд), для этого на оси ординат откладывают Ах – оптическую плотность исследуемого раствора без добавки, на оси абсцисс откладывают значения концентраций добавок, приняв за условный ноль концентрацию раствора без добавки. Из точек Сд1, Сд2, Сд3 восстанавливают перпендикуляры, на которых откладывают значения оптических плотностей растврв с добавками: Ах+д1, Ах+д2, Ах+д3. Через полученные точки точки проводят прямую. Экстаполяция прямой до пересечения с осью абсцисс дает отрезок ОСх, величина которого соответствует искомому содержанию железа.
Для уменьшения погрешности определения необходимо, чтобы первая добавка была близка к искомой концентрации, а вторая – в два раза больше первой, в этом случае обеспечивается угол наклона прямой, близкий к 45о.
Работа 4. Определение меди в виде аммиаката методом дифференциальной фотометрии. Метод основан на образовании комплексного соединения ионов меди(II) с аммиаком, обладающего интенсивной сине-фиолетовой окраской. Окраска аммиаката меди обусловлена d→d* переходами вследствие расщепления основного электронного состояния ионов меди в поле лигандов. Процесс взаимодействия ионов меди с аммиаком носит ступенчатый характер: Cu2+ + NH3 ↔ [Cu(NH3)]2+; Cu2+ + 2 NH3 ↔ [Cu(NH3)2]2+; Cu2+ + 3 NH3 ↔ [Cu(NH3)3]2+; Cu2+ + 4 NH3 ↔ [Cu(NH3)4]2+. Так как устойчивость образующихся комплексов различается мало (lg β1=3.99; lg β2=7.33; lg β3=10.61; lg β4=12.03), то в растворе будет находиться смесь нескольких аммиакатов меди, количественное соотношение которых зависит от концентрации аммиака, присутствующего в растворе, что иллюстрируется данными диаграммы:
Для аналитических целей необходимо выбрать такую концентрацию аммиака, при которой в растворе будет преобладать один из комплексов, наиболее эффективно это возможно при lg [NH3] =1. Молярный коэффициент светопоглощения тетрааммиаката меди при λ=640 нм равен 1∙102. Низкое значение ε позволяет определять достаточно высокие концентрации ионов меди. Для повышения воспроизводимости определения используют метод дифференциальной фотометрии, когда раствор сравнения содержит представляет собой раствор, содержащий ионы меди в виде аммиаката, точно известной концентрации. Определению меди в виде аммиачного комплекса мешают ионы металлов, образующие окрашенные комплексы аналогичного типа, например, кобальт и никель, или малорастворимые гидроксиды железа, свинца и алюминия. Для устранения мешающего действия этих элементов применяют маскирующие комплексообразователи. Приборы и реактивы: фотоэлектроколориметр КФК-2; рабочий раствор соли меди, содержащий 1 мг/мл Cu2+; для приготовления рабочего раствора навеску 3.931 г CuSO4 ∙ 5H2O растворяют в 25 мл 2м раствора H2SO4, доводят объем раствора до 1.00 л дистиллированной водой; аммиак, 10%-ный раствор. Приготовление стандартных растворов. Готовят шесть стандартных растворов, содержащих 2.5; 5.0; 7.5; 10.0; 12.5; 15.0 мг меди в 50.0 мл. Для этого в мерные колбы вместимостью 50.0 мл переносят рабочий раствор соли меди, добавляют в каждую колбу 10.0 мл 10% -ного раствора аммиака и доводят объем каждого раствора до 50.0 мл дистиллированной водой. Через 10 минут приступают к измерению оптической плотности. Выполнение работы. Выбор светофильтра. Раствор, имеющий окраску средней интенсивности, фотометрируют относительно воды со всеми светофильтрами поочередно, записывая результаты в таблицу:
По результатам измерений строят график: А=f(λ). Для дальнейшей работы выбирают светофильтр, соответствующий наибольшему значения светопоглощения исследуемого раствора. Посторенние градировочного графика. С выбранным светофильтром поочередно фотометрируют стандартные растворы относительно раствора, содержащего 5.0 мг меди. Если содержание меди в фотометрируемом растворе меньше, чем в растворе сравнения, применяют обратный порядок измерений: фотометрируемый раствор условно принимают за раствор сравнения, устанавливают по нему оптический ноль и по отношению к нему измеряют светопоглощение исследуемого раствора. Найденное значение поглощения берут со знаком «минус». Сочетание прямого (Со>Сх) и обратного (Со<Сх) порядков измерений в дифференциальном методе называют двусторонним дифференцированием. Определение содержания меди(II) в растворе. К анализируемому раствору, содержащему соль меди (II), приливают 10.0 мл 10%-ного раствора аммиака и доводят объем раствора до 50.0 мл дистиллированной водой. Приготовленный раствор через 10.0 мин фотометрируют с выбранным светофильтром относительно выбранного раствора сравнения, содержащего 5.0 мг меди, используя при необходимости приемы двустороннего дифференцирования. Пользуясь градуировочным графиком, находят содержание меди в анализируемом растворе.
Хроматографические методы.
Хроматографией называют метод разделения веществ, в котором разделяемые вещества распределяются между неподвижной и подвижной (движущейся) фазами. В качестве неподвижной фазы используют твердое вещество или жидкость, нанесенную на твердый носитель. Хроматографическое разделение основано на том, что отдельные компоненты образца перемещаются по колонке (или листу фильтровальной бумаги) с различной скоростью и соответственно за одно и то же время проходят различные отрезки пути. Растворитель, проходящий через колонку, называют элюентом, а процесс перемещения вещества вместе с элюентом – элюированием. В современной колоночной хроматографии твердую фазу, как правило, не извлекают из колонки и разрезают на полосы. Вместо этого проводят элюирование до тех пор, пока компоненты образца не выйдут один за другим из колонки. Для определения компонентов также можно использовать детектор, установленный на выходе из колонки. Показания такого детектора можно регистрировать автоматически. Полученную кривую называют хроматограммой. Каждый пик на такой кривой соответствует отдельному компоненту образца, а площадь пика характеризует относительное содержание данного компонента. Современные хроматографические методы разделения имеют исключительно большие возможности. В благоприятных случаях в течении нескольких минут можно полностью разделить до 30 компонентов, содержащихся в одном образце. Ионообменная хроматография В основе ионообменной хроматографии лежит обратимый стехиометрический обмен ионов, содержащихся в хроматографируемом растворе, на подвижные ионы веществ, называемых ионитами или ионообменниками. Ионит представляет собой высокополиллорную матрицу, которая обладает положительным или отрицательным зарядом, компенсирующимся ионами противоположного знака, так что в целом ионит нейтрален. Если ионит, содержащий противоион только одного вида А, поместить в раствор, в котором находятся ионы другого вида В, то ионы А будут покидать ионит и переходить в раствор, а ионы В будут строго в эквивалентном количестве переходить в ионит. При достижении равновесия ионит и раствор будут содержать ионы А и В в определенном соотношении, определяемом константой ионообменного равновесия. К числу важнейших свойств ионитов относят их обменную емкость. Обменная емкость, определяемая в статических условиях, может отличаться от величины, полученной в динамических условиях. Последняя характеризуется двумя показателями: динамической обменной емкостью до проскока (ДОЕ) и полной динамической обменной емкостью (ПДОЕ). ДОЕ представляет собой емкость ионита, определяемую до появления данного иона в вытекающем из колонки раствора. ПДОЕ определяется по полному прекращению извлечения данного иона из раствора. Ионообменная хроматография нашла широкое применение в аналитической практике. Определение солей. Большинство растворимых солей можно определить используя катионообменную смолу в Н+– форме Z RH++MAz=Z HA+RzMz+ Содержание катиона в образце определяют титрованием образующей кислоты HA. Деионизация. Ионный обмен– обычный метод получения деионизированной воды. Очищаемую воду пропускают через колонку, содержащую смесь катионита в Н+– форме и анионита в ОН—форме. При этом примеси катионов, содержащихся в воде, замещаются на ионы водорода, а примеси анионов соответственно на Гидроксид–ионы. В итоге практически все примеси удаляются из воды. Замещение мешающих ионов. Ионный обмен часто используют для замены нежелательных ионов на другие, не мешающие аналитическому определению. Например, ионы Fe (III) и калия мешают определению сульфат–иона, соосаждаясь сульфатом бария. Для устранения их влияния анализируемый раствор пропускают через катионит в Н+– форме и мешающие примеси обмениваются на ионы водорода. Таким же образом удаляют фосфат –ионы, которые мешают определению кальция и некоторых других ионов. В этом случае раствор пропускают через анионит с Cl–-форме. подготовка ионита к работе. Для анализа необходимо применить иониты одного зернения. Соотношение диаметра колонки и диаметра отдельного зерна должно быть не менее 40:1. Можно рекомендовать следующие размеры колонок в зависимости от размеров зерен ионита:
В качестве колонок рекомендуется применять стеклянные трубки с краном внизу. Важной стадией работы с ионообменниками являются их подготовка к эксперименту и регенерация после обработки. Товарные иониты представляют собой смолы, в которых наряду с основным веществом– высокомолекулярной смолой–присутствуют низкомолекулярные фракции, различные примеси, особенно ионы железа. Кроме того, ионит должен быть доведен до набухания, т.к. только в набухшем состоянии его следует загружать в колонку. Подготовка катионита. Товарный образец катионита измельчают, просеивают и для подготовки отбирают фракцию с величиной зерен 0,5-0,25мм. Порцию этой фракции катионита помещают в химический стакан и заливают 5–кратным по объему количеством насыщенного раствора NaCl и оставляют для набухания на 24 часа, после чего раствор декантируют, а катионит переносят в делительную воронку, в которой его промывают не менее 5 раз 5%-ным раствором HCl. Затем катионит промывают дистиллированной водой до нейтральной реакции по метилоранжу. Обработка катионита раствором HCl переводит его в Н+–форму. Отмытый от кислоты катионит отделяют, просушивают на фильтровальной бумаге до такого состояния, чтобы зерна свободно отделялись друг от друга. Подготовленный таким образом катионит хранят в банке с притертой пробкой и используют по мере надобности. Подготовка анионита. Анионит, как и в случае с катионитом, измельчают, просеивают и отбирают фракцию с размером зерен 5,5-0,25 мм, обрабатывают насыщенным раствором NaCl. Затем анионит обрабатывают 2 % раствором HCl до полного удаления ионов Fe 3+ (проба с KSCN). После этого анионит промывают 10-кратным дистиллированной воды, 5%–ным раствором щелочи, а затем 10%–ным раствором щелочи до полного удаления ионов хлора(проба с AgNO3). Заканчивают подготовку анионита промыванием дистиллированной водой до нейтральной реакции по фенолфталеину. Подготовленный таким образом анионит хранят под водой в банке с притертой пробкой. Только непосредственно перед загрузкой анионит вынимают из банки, отжимают между листами фильтровальной бумаги и подсушивают на воздухе. В таком виде анионит загружают в колонку. 3.1.2. Определение физических свойств ионитов. Иониты характеризуются рядом физико–химических и физико–механических свойств. К физическим свойствам относятся: влажность, фракционный состав в набухшем состоянии, насыпной вес, удельный объем, набухаемость. Приборы и реактивы. 1. Цилиндры мерные на 10, 100, 250 мл. 2. Сушильный шкаф, эксикатор. 3. Бюксы. 4. Катионит в Н–форме, анионит в ОН–форме или Cl–форме.
Выполнение работы. а) Определение влажности ионита. В бюкс с притертой пробкой помещают навеску 3-4 г катионита, взвешенного с точностью до 0,002г. Катионит высушивают до постоянной массы при 1050С. Перед взвешиванием бюкс с катионитом охлаждают в эксикаторе в течении 1 часа. Расчет влажности ионита проводят по формуле: ω= m1– масса ионита до сушки, г m2– масса ионита после сушки, г ω–влажность, %. Определение влажности низкоосновных анионитов выполняется по той же методике при t=75–800С б) Определение насыпного веса анионита. В мерный цилиндр объемом 10 мл вносят 5 г предварительно подготовленного ионита. Встряхивают пробу, постукивая дном цилиндра по твердой поверхности до прекращения осадки слоя ионита. Насыпной вес ионита вычисляют по формуле: В= В– насыпной вес ионита, г/мл; m–масса ионита, г; V– объем ионита, мл. в) Определение набухаемости ионита. Под набухаемостью ионита подразумевают изменение удельного объема при переходе его из одной формы в другую. Набухаемость представляет разницу удельных объемов набухшего и сухого ионита и выражается в мл/г (V0–V׳0). Под удельным объемом ионита понимают отношение объема набухшего ионита к массе сухого (
Определение обменной емкости ионита. Емкость ионообменной смолы определяется количеством ионов, обмениваемых одним граммом смолы. Емкость сухой сульфированной катионнообменной смолы в Н+– форме обычно равна примерно 5 мэкв/г, емкость влажной смолы–1,8 мэкв/мл. Емкость сухого анионита, находящегося в Сl—форме, равна 3,0–3,5 мэкв/г, а емкость влажной смолы равна 1,2 мэкв/мл. Различают: 1) ионную обменную емкость, которая определяется количеством активных ионогенных групп, входящих в состав ионообменной смолы, и является постоянной величиной, соответствующей состоянию предельного насыщения всех способных к ионному обмену активных групп. 2) обменную емкость по отдельным отношенным группам, которая соответствует предельному насыщению активных групп только одного типа и также является постоянной величиной; 3)равновесную обменную емкость, которая зависит от многих факторов, определяющих равновесие, и является переменной величиной. Наиболее распространенными реакциями, лежащими в основе определения емкости статическим методом, являются следующие: RH+NaOH=RNa+H2O; для катионообменных смол 2 RH+CaCl2=R2Ca+2HCl 2 RNa+ CaCl2= R2Ca+2NaCl
R` OH+HCl= R`Cl+ H2O; для анионообменных смол 2 R`Cl+H2SO4= R2SO4+2 HCl R` OH+NaCl= R`Cl+NaOH
Выполнение работы Берут две навески (по 1 г) предварительно подготовленного ионита с известной влажностью и помещают в конические колбы на 200 мл. Навески берут с точностью до 0,01 г. Затем в одну колбу помещают точно 100 мл 0,1 н CaCl2 , в другую 100 мл 0,1 н раствора NaOH, после чего колбы закрывают и оставляют стоять в течении 10 часов. Периодически содержимое колб осторожно взбалтывают. Затем из них отбирают по 25 мл раствора и титруют в присутствии метилоранжа первый раствор 0,1 н раствором NaOH, второй–0,1 н раствором HCl. Расчет производится по формуле: СОЕ CaCl2= Где а– навеска катионита,г. Для данного случая СОЕ CaCl2 =
СОЕ NaOH=
Определение ОЕ ионитов в динамических условиях (ДОЕ и ПДОЕ) Реактивы: 1. Cu(NO3)2 0.05 н раствор. 2. H2SO4, 2 н раствор 3. KJ, 20%-ый раствор; 4. Na2S2O3, 0,1 н раствор; 5. крахмал, 1%-ный раствор 6. смола в Н+–форме.
Выполнение работы Берут навеску катионита 5 г, помещают в колонку и пропускают через неё 0,05 н раствор соли меди со скоростью 1 капля в секунду. Вытекающий из колонки раствор собирают в мерные колбы на 25 мл (первые 1,0-1,5 мл раствора отбрасывают). В каждой порции вытекающего раствора определяют количество меди иодометрически. Для этого раствор из мерной колбы количественно переносят в колбу для титрования, добавляют 4 мл 2 н H2SO4, 2 г KJ или 10 мл 20%-ого раствора иодида калия и после которого выдерживают раствор в темноте, выделившийся иод оттитровывают 0,1 н раствором Na2S2O3 до желтой окраски. Затем добавляют 2-3 крахмала и продолжают титровать до обесцвечивания синей окраски. Пропускание раствора соли меди через колонку продолжают до тех пор, пока концентрация вытекающего из колонки раствора не станет равной концентрации исходного раствора. По разности титров исходного раствора и фильтратов можно определить количество ионов меди поглощенное сорбентом из каждой порции. Суммируя эти количества по всем порциям и деля полученную величину на навеску ионита, получим значение полной обменной емкости (ПДОЕ). Количество ионов меди, поглощенное ионитом, рассчитывается исходя из всего объема фильтрата, не содержащего ионов меди, и исходной концентрации меди в растворе. Для определения обменной емкости до проскока это количество ионов меди делят на взятую навеску катионита.
ДОЕ= Где Сн(Cu)- нормальность исходного раствора соли меди. V- объем фильтрата, не содержащего меди а- навеска ионита, г ПДОЕ= Где Сн(Сu)- нормальность соли меди V Cu- объем соли, прошедшей через колонку до полного насыщения ионами меди Сн (Na2S2O3)- нормальность тиосульфата натрия V Na2S2O3- общий объем тиосульфата натрия пошедший на титрование аликвотных частей соли меди после прохождения через колонку. а) навеска ионита По данным опыта строят выходную кривую в координатах:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-28; просмотров: 1055; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.141.29.165 (0.011 с.) |

 Фотоэлектроколориметр КФК-2МП.
Фотоэлектроколориметр КФК-2МП.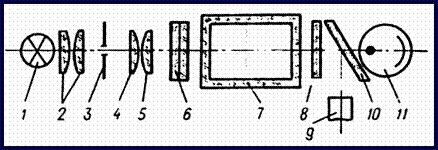
 ,
, .
.



 , где
, где , где
, где мг/г). Относительная набухаемость определяется как отношение удельного объема в одних условиях к удельному объему в других.
мг/г). Относительная набухаемость определяется как отношение удельного объема в одних условиях к удельному объему в других.


 ,
, ,
,


