
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Гомогенный катализ в газовой фазе
Примером катализа этого типа может служить каталитическое разложение оксида диазота N2O в газовой фазе. Оксид диазота-распространенное анестезирующее средство, известное под названием «веселящий газ». При комнатной температуре он сравнительно инертен, разлагается только при температурах выше 1000 К:
Однако его разложение катализируется следами газообразного хлора, особенно в присутствии света. По-видимому, роль катализатора выполняют радикалы хлора, которые образуются в результате фотолиза газообразного хлора (см. выше). Предположительно, радикал хлора реагирует с N2O, образуя промежуточный радикал QO":
Одним из примеров каталитических реакций в растворах является распад перекиси водорода в воде в присутствии ионов йода:
В водных растворах распространено явление кислотно-основного катализа. Еще в конце XIX в. было установлено влияние добавок кислот и оснований на скорость реакций в водных растворах. Это привело к заключению, что ионы водорода и гидроксила отличаются каталитическими свойствами. Дальнейшие исследования показали, что каталитическая активность кислот и оснований сохраняется и в неводных растворах, где электролитическая диссоциация слаба. Это явление может быть понято на основе представлений Бренстеда и Лаури о кислотах и основаниях как о веществах, способных обмениваться протонами. Соответственно кислотой называется соединение, способное отдавать протоны, т.е. являющееся "донором" протонов, а основанием - "акцептор" - вещество, способное присоединять протоны.
Например, в реакции
+ = +
молекула воды является кислотой, отдающей протон, а аммиак - основанием, принимающим протон. При этом образуется новая кислота NH
и новое основание ОН-. Подобные реакции, заключающиеся в переходах протона, могут происходить и в неводных растворах, например, в жидком аммиаке: Равновесие, устанавливающееся в таких реакциях, называют кислотно-основным. В общем виде оно изображается уравнением K + O = K+ + O-, где K, О - исходные кислота и основание; K+, О- - образующиеся. Согласно изложенным выше представлениям катализ кислотой обусловлен переходом протона от К к исходному веществу R с образованием неустойчивого промежуточного соединения R H+, которое, распадаясь, дает конечный продукт X и регенерирует кислоту:
R + K = R H+ + O- и R H+ + O- → X + K. Соответственно схема катализа основаниями имеет вид: R H + O- = R - + K и R - + K → X + O-. Кинетику таких каталитических реакций, например, с участием кислоты, можно рассматривать как последовательный, процесс, протекающий в две стадии:
где k 1, k 2 и k 3 - константы скоростей соответствующих реакций. Комплекс R H+ можно рассматривать как переходное состояние, находящееся в равновесии с исходным веществом R. На первой стадии при таком равновесии скорости прямой ω→ и обратной ω← реакций R + K ←→ R H+ → O- равны между собой, т.е. ω→ = k 1 cRc K = ω← = k 2 cR H+ c O-2. Отсюда величина CRH+ = (ki/k2)cKcK/c0- и, следовательно, скорость суммарной реакции выражается уравнением: -
= k 3 cR H+ = k 3(k 1/ k 2) cRc K/ c O-. (XIX.1) Таким образом, при постоянном содержании исходного вещества скорость реакции пропорциональна концентрации катализатора. Если же скорость распада промежуточного соединения R H+ с образованием X (в данном случае О-) намного больше скорости его обратного превращения в R, то между R и R Н+ не может установиться равновесие. В этом случае суммарная скорость процесса определяется скоростью первой стадии (перехода протона от кислоты к R), и, поскольку концентрация кислоты К остается постоянной, должна иметь место реакция первого порядка с константой скорости k = k 1[K].
25. Гетерогенный катализ. Характеристика твердых катализаторов. При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель. Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы. 1. Диффузия реагирующих веществ к поверхности твердого вещества 2. Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их
3. Химическая реакция между реагирующими молекулами 4. Десорбция продуктов с поверхности катализатора 5. Диффузия продукта с поверхности катализатора в общий поток Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод). Течение реакции именно на поверхности катализатора можно продемонстрировать на опыте, в котором пластинку из платины нагревают в пламени газовой горелки, затем пламя тушат и пускают на пластинку струю газа из горелки, при этом пластинка снова раскаляется докрасна — окисление метана происходит на поверхности металла[3]. Твердые катализаторы — это, как правило, высокопористые вещества с развитой внутренней поверхностью, характеризующиеся определенной пористой и кристаллической структурой, активностью, селективностью и рядом других технологических характеристик. Рассмотрим некоторые характеристики твердых катализаторов.
• Температура зажигания — это минимальная температура, при которой технологический процесс начинает идти с достаточной для практических целей скоростью. • Селективностью или избирательностью катализатора называют его способность избирательно ускорять целевую реакцию при наличии нескольких побочных. • Селективность зависит не только от выбранного катализатора, но и от условий проведения процесса, области протекания гетерогенно-каталитического процесса (кинетической, внешне- или внутреннедиффузионной) и т. д. Пористая структура характеризуется размерами и формой пор, пористостью (отношением свободного объема пор к общему объему), удельной поверхностью катализатора Распределение пор по размерам может оказаться таким, что часть поверхности катализатора окажется недоступной для реагирующих молекул большого размера и, кроме того, скорость превращения реагентов в конечные продукты может уменьшаться вследствие затруднения диффузии реагентов внутри пор. Промотирование и отравление катализаторов. Часто введение очень небольшого количества (долей процента) какой-либо посторонней добавки к. основному катализатору приводит либо к резкому повышению его активности, либо, наоборот, к снижению активности на несколько порядков. В первом случае говорят о промотировании, во втором — об отравлении катализатора
Добавки могут вступать с основным катализатором в химическое взаимодействие, образуя на поверхности продукты, обладающие более высокой каталитической активностью. Они могут изменить условия взаимодействия с реагентами в местах контакта основного компонента и промотора, а также увеличить дисперсность или стабилизировать пористую и кристаллическую структуру катализатора 1-я стадия - д иффузия реагента к внешней поверхности катализатора. Эта стадия назвается стадией внешней диффузии 2-я стадия - диффузия внутри пор катализатора (стадия внутренней диффузии). 3-я стадия – адсорбция. Различают физическую адсорбцию и хемосорбцию. При хемосорбции молекулы адсорбата образуют поверхностные соединения с адсорбентом. 4-я стадия - химическая реакция, механизм этой реакции может быть различным, от него зависит вид кинетического уравнения. В результате поверхностной реакции образуется адсорбированный продукт.
5-я стадия - десорбция продукта с поверхности катализатора. 6-я стадия. - диффузия из пор к внешней поверхности катализатора 7-я стадия. - диффузия от поверхности катализатора Стадии 3, 4, 5 являются центральными в ходе каталитического процесса. Суммарно их можно рассматривать как поверхностную химическую реакцию. Эти стадии могут протекать одновременно с предыдущими — диффузионными — стадиями, причем как на внешней поверхности зерна катализатора, так и, в основном, на внутренней поверхности пор. 6-я стадия. Десорбированные газообразные продукты диффундируют из пор к внешней поверхности катализатора (обратная внутренняя диффузия). 7-я стадия. Газообразные продукты диффундируют от поверхности катализатора в тазовый поток через пограничную пленку, окружающую зерно катализатора. Таким образом, гетерогенно-каталитический процесс — это сложная система последовательных и параллельных стадий, имеющих разную природу. Как и в случае некаталитического гетерогенного процесса, одна из стадий может оказывать наиболее сильное тормозящее воздействие на весь процесс, тогда скорости остальных стадий «подстраиваются» под скорость этой наиболее затрудненной стадии, которая может быть названа лимитирующей. 26. Термодинамика электрохимических процессов. Электродное равновесие. Уравнение Нернста.
Процессы взаимного превращения химической и электрической форм энергии называют электрохимическими процессами. Электрохимические процессы можно разделить на две основные группы: 1) процессы превращения химической энергии в электрическую (в гальванических элементах); 2) процессы превращения электрической энергии в химическую (электролиз). Простейшая электрохимическая система состоит из двух электродов и ионного проводника между ними. Электроды замыкаются металлическим проводником. Ионным проводником (проводником 2-го рода) служат растворы или расплавы электролитов, а также твердые электролиты. Электродами называют проводники, имеющие электронную проводимость (проводники 1-го рода) и находящиеся в контакте с ионным проводником. Для обеспечения работы системы электроды соединяют друг с другом металлическим проводником, называемым внешней цепью электрохимической системы.
При погружении металла в раствор начинается сложное взаимодействие металла с компонентами раствора. В результате взаимодействия происходит окисление металла и его гидратированные ионы переходят в раствор, оставляя в металле электроны, заряд которых не скомпенсирован положительно заряженными ионами в металле: М тH2О = М(Н2О)mn пе. Металл становится заряженным отрицательно, а раствор -положительно. Положительно заряженные ионы из раствора притягиваются к отрицательно заряженной поверхности металла. На границе металл-раствор возникает двойной электрический слой. Между металлом и раствором возникает разность потенциалов, которая называется электродным потенциалом или потенциалом электрода. По мере перехода ионов в раствор растет отрицательный заряд поверхности металла и положительный заряд раствора, что препятствует окислению металла. Наряду с этой реакцией протекает и обратная — восстановление ионов металла до атомов: Равновесие является динамическим. Процессы при равновесии идут с одинаковой скоростью в прямом и обратном направлениях. Потенциал, устанавливающийся в условиях равновесия электродной реакции, называется равновесным электродным потенциалом. Абсолютные значения электродных потенциалов экспериментально определить невозможно. Однако можно определить разность электродных потенциалов. Поэтому для характеристики электродных процессов пользуются относительными значениями электродных потенциалов. Для этого находят разность потенциалов измеряемого электрода и электрода, потенциал которого условно принимают равным нулю. Схематически электроды записываются в виде Me/Men , a электродный потенциал электрода - Активность металлов в окислительно-восстановительном электрохимическом процессе «металл-ион металла», а следовательно, и величины электродного потенциала, зависят от природы
2,3 - коэффициент натурального логарифма в десятичный; R - универсальная газовая постоянная, равная 8,314 Дж/моль-К; Т- температура, равная 298 К; n - число электронов, участвующих в данной электродной реакции; F- число Фарадея, равное 96500 Кл/моль;
После подстановки значений постоянных величин уравнение Нернста (7.1) приобретает следующий вид:
Значения стандартных электродных потенциалов (j0) определяют относительно стандартного водородного электрода, потенциал которого принят за нуль, т. е. j02H /H2 = 0.
27. Электрохимический ряд напряжения металлов. Классификация электродов. Металлы, расположенные в порядке возрастания алгебраического значения их стандартного электродного потенциала, составляют электрохимический ряд напряжений. Потенциал каждого электрода (металла) зависит от природы металла, концентрации его ионов в растворе, температуры. Разность потенциалов между металлом, погружённым в раствор своей соли с концентрацией ионов металла 1 моль/л, и стандартным водородным электродом при стандартных условиях называется стандартным электродным потенциалом металла (E0). Электродные потенциалы щелочных и щелочноземельных металлов рассчитываются теоретически, так как эти металлы в водных растворах взаимодействуют с водой. Значение электродного потенциала количественно характеризует способность металла отдавать электроны, то есть его восстановительные свойства (химическую активность металла). В этом ряду восстановительная активность металлов в водных растворах слева направо уменьшается: металлы, стоящие в начале ряда, легко отдают электроны и превращаются в положительно заряженные ионы; металлы, стоящие в конце ряда, с трудом отдают электроны. И наоборот, окислительная способность катионов металлов слева направо увеличивается Металлический литий Li – самый сильный восстановитель, а золото Au – самый слабый. Ион золота Au3+ – самый сильный окислитель, ион лития Li+ – самый слабый. На основании ряда напряжений можно сделать некоторые важные заключения о химической активности металлов. 1. Каждый металл вытесняет из солей другие металлы, имеющие большие (уд. на 1-ый слог) значения стандартных электродных потенциалов, то есть являющиеся менее сильными восстановителями. 3. Металлы, имеющие очень низкие стандартный электродный потенциал меньше нуля (то есть потенциала стандартного водородного электрода), способны вытеснять водород из кислот. 4. Металлы, имеющие очень низкие значения стандартного электродного потенциала, то есть являющиеся сильными восстановителями (от лития до натрия) в любых водных растворах взаимодействуют прежде всего с водой. Классификация электродов. Различают электроды, обратимые относительно катиона и обратимые относительно аниона. Примером первых может служить цинковый и медный электроды в растворе солей. Примером вторых – хлорный и кислородный электроды. В основу классификации электродов положено агрегатное состояние и растворимость окисленной и востановленной формы веществ, участвующих в электродной реакции. Электроды делятть на электроды первого и второго рода, газовые амальгамные и окислительно-востановительные. Особым видом электродов является стеклянный электрод, в котором скачет потенциала возникает из-за ионнообменной адсорбции. Электроды первого рода. К ним относятся металл-ионные и неметалл-ионнные электроды. Первые обратимы относительно катионов. Равновесие устанавливается между металлом, активность которого величена постоянная, и его катионами, активность которых можно менять в широких пределах.
Электроды второго рода. В них окисленная форма вещества берется в виде труднорастворимого соединения(Соль, гиброксид, оксид), которе находится в электролите с одноименным анионом. Электролит необходим для уменьшения сопростивления раствора, а также для поддердивания постоянной концентрации катионов, относительно которых обратим электрод. На электродах второго рода устанавливается равновесие:
Газовые электроды. Примером может служить водородный электрод. Для приготовления газовых электродов берут платину, которая не посылает свои ионы в раствор и обладает высокойадсорбционной способностью. На границе платина –раствор устанавливвается равновесие. Потенциал газовых электродов зависит как от активности востановленной, так и окисленной формы вещества. Концентрация газооразного вещества по закону Генри прямопропорциональна его давлению над раствором. Поэтому в формулу Нернста вместо его концентрации подствляют парциальное давление. Уравнение Нернста для потенциала водродного электрона имеет вид Амальгамные электроды. Многие металлы (медь середьро золото кадмиц цинк) растворяются в ртути. А амальгаме активность растворенного металла уже не равно единице, в связи с чем потенуиал амальгамного электрода зависит от активности металла и его катионов. Активность металла не равно его атомной доле в амальгаме вследствие того, что ртуть химически взаимодействует с металлом. Поэтому стандартные потенциалы амальгамного и металл ионного электродов не равны. Амальгамные электроды находят широкое применение в полярографии при производстве щелочей и хлора, а гальванических элементах. Оскислительно-востановит электроды.(Редокси-элетроды)К ним относят полуэлементы в которых окисленная и вост форма вещества находится в растворе. Элеттродная реакция осуществяется на инертном электроде(использ. Платину.)
Возникает второй скачок потенциала
Электрохимическая кинетика - раздел теоретической электрохимии, рассматривающий закономерности, которым подчиняется скорость электродных процессов. Электрический ток, проходящий через границу электрод - ионная система, связан с протеканием электродного процесса (фарадеевский ток) и с заряжением двойного электрического слоя (ток заряжения). Если свойства поверхности электрода не изменяются во времени, протекающий через электрод ток определяется только скоростью самого электродного процесса и размерами электрода. В этих условиях плотность тока i служит мерой скорости электрохимической реакции. Если электрод находится при равновесном потенциале Ер, ток i = 0. При пропускании через электрод электрического тока потенциал электрода отклоняется от Ер на величину ΔE которая называется поляризацией электрода. Для величины ΔE часто используют термин "перенапряжение" (обозначение η). Поляризация электрода обусловлена конечной скоростью электродного процесса, а потому она является функцией плотности тока. Функциональная зависимость ΔE от i (или i от ΔE) называется поляризационной характеристикой электрода. Задача электрохимической кинетики заключается в установлении общих закономерностей, которым подчиняются поляризационные характеристики электродов, с целью регулирования скорости электродных процессов. Решение задач электрохимической кинетики имеет большое практическое значение, поскольку уменьшение поляризации ΔE при заданной плотности тока позволяет существенно повысить кпд использования электрохимических систем. Электрохимическая кинетика является теоретической основой электрохимической защиты металлов от коррозии. Поскольку электродные процессы являются гетерогенными и состоят из ряда последовательных стадий, общая поляризация ΔE определяется совокупностью поляризаций ΔEj соответствующих отдельным стадиям. Стадия, дающая наибольший вклад в суммарную величину ΔE является лимитирующей, она определяет вид поляризационные характеристики. Чтобы определить лимитирующую стадию, сравнивают закономерности исследуемого электродного процесса с закономерностями, характерными для различных стадий. Определение лимитирующей стадии позволяет, меняя условия, изменить скорость электродного процесса в нужном направлении. Напряжение разложения Минимальная разность потенциалов, которую нужно создать между электродами, чтобы начался электролиз, называется напряжением разложения электролита. Эта величина равна сумме потенциалов разряда ионов на электродах. В отсутствии перенапряжения на электродах напряжение разложения равно сумме равновесных потенциалов электродов, образующихся после начала электролиза. Величина напряжения разложения более или менее точно определяется для электролита при заданных условиях лишь в случае выделения на электродах индивидуальных твердых веществ, например, чистых металлов. Если в результате электролиза образуются твердые или жидкие растворы и, особенно, газы, напряжение разложения зависит от формы и размеров электродов, характера их поверхности, условий отвода газов и др. Таким образом, напряжение разложения данного электролита не является однозначной характеристикой и зависит от условий проведения электролиза В растворах кислот выделение водорода происходит по схеме 2Н3О++ 2е®2Н2О + Н2, т.е. разряжаются ионы гидроксония. В растворах щелочей также возможен их разряд, но в таких растворах их концентрация очень мала. Таким образом, при электролизе растворов кислот, щелочей, а также соответствующих солей щелочных и щелочноземельных металлов на электродах протекает первичный процесс разложения воды. Роль остальных ионов сводится к обеспечению достаточной электропроводности растворов. Для растворов других веществ ЭДС поляризации отличаются, что указывает на различный характер электродных процессов для разных веществ. В растворах солей металлов, менее электроотрицательных, чем водород, на катоде может выделяться металл. При электролизе растворов кислот, молекулы которых не содержат кислород, на аноде обычно разряжаются анионы. Интересно в этом отношении поведение соляной кислоты: в концентрированных растворах на аноде выделяется хлор, а в разбавленных – кислород, при этом изменяется ЭДС поляризации. Это связано с тем, что при разбавлении кислоты уменьшается активность хлорид-ионов. Равновесный потенциал хлорного электрода становится более положительным, чем потенциал разряда ионов ОН–, поэтому изменяется анодный процесс – уменьшается разряд ионов хлора и происходит разряд ионов гидроксила или молекул воды. Перенапряжение Во многих случаях для того, чтобы начался электролиз, к электролитической ванне необходимо приложить напряжение, превосходящее на некоторую конечную величину ЭДС электрохимической поляризации. Разность между напряжением разложения и суммой равновесных потенциалов электродов называется перенапряжением. Величина перенапряжения на электроде зависит от природы электрода, состава раствора, плотности тока и других факторов. Перенапряжение в целом в электролитической ванне равно тому избыточному напряжению, которое нужно приложить к ванне сверх ее равновесной ЭДС, чтобы начался электролиз. Избыточное напряжение включает, кроме перенапряжения на электродах, также падение напряжения на внутреннем сопротивлении, которое зависит от электрического сопротивления раствора Реакция выделения водорода имеет большое практическое значение во многих электрохимических процессах – электролиз воды и различных водных растворов, хлорный электролиз, работа химических источников тока, процессы коррозии и др., поэтому перенапряжение водорода изучено наиболее подробно. При выделении водорода из растворов минеральных кислот и оснований, а также из водных растворов солей при малых отклонениях от равновесного потенциала наблюдается линейная зависимость между перенапряжением и плотностью тока. С увеличением плотности тока зависимость выражается уравнением Тафеля и для некоторых металлов постоянные а и b сохраняют свое значение до высоких плотностей тока. Перенапряжение водорода снижается с повышением температуры, при этом температурный коэффициент зависит от природы металла и плотности тока.
|
|||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-16; просмотров: 628; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.12.205 (0.054 с.) |




 Величина электродного потенциала определяется в вольтах (В).
Величина электродного потенциала определяется в вольтах (В).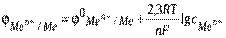 электродов, концентрации ионов в растворе, рН раствора, давления, температуры и определяются уравнением Нернста:
электродов, концентрации ионов в растворе, рН раствора, давления, температуры и определяются уравнением Нернста: - электродный потенциал определяемого электрода, В;
- электродный потенциал определяемого электрода, В; - стандартный электродный потенциал, определяемого электрода, возникающий на границе электрод-электролит при концентрации ионов в растворе, равной 1 моль/дм3, в стан дартных условиях (Т = 298 К, р = 100 кПа), В;
- стандартный электродный потенциал, определяемого электрода, возникающий на границе электрод-электролит при концентрации ионов в растворе, равной 1 моль/дм3, в стан дартных условиях (Т = 298 К, р = 100 кПа), В;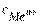 - молярная концентрация ионов в растворе электролита, моль/дм3.
- молярная концентрация ионов в растворе электролита, моль/дм3. (7.2)
(7.2)
 Т.О. потенциал электродов второго рода зависит от активности анионов в растворе и электроды как бы обратимы относительно аниона. Электроды второго рода применяют при измерении потенциалов отдельных электродов в качестве электродов сравнения вместо стандартного водородного электрода.
Т.О. потенциал электродов второго рода зависит от активности анионов в растворе и электроды как бы обратимы относительно аниона. Электроды второго рода применяют при измерении потенциалов отдельных электродов в качестве электродов сравнения вместо стандартного водородного электрода. Платина катализирует приведенные элекктродные реакции, что обеспечивает достаточно высоких ток обмена. С целью увеличения тока обмена увеличивают поверхность платины, покрывая ее платиновой чернью которую наносят на электролизом Н2PtCl6.
Платина катализирует приведенные элекктродные реакции, что обеспечивает достаточно высоких ток обмена. С целью увеличения тока обмена увеличивают поверхность платины, покрывая ее платиновой чернью которую наносят на электролизом Н2PtCl6.





