Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Молекулярные механизмы опухолевого ростаСодержание книги
Поиск на нашем сайте МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ ОПУХОЛЕВОГО РОСТА
Учебное пособие для студентов медицинских вузов Под ред. Б.А. Фролова
Оренбург
УДК 616-006. (075.8) + 616 – 092 ББК 55.6 я 7
Авторы: Б.А. Фролов - докт. мед. наук, профессор, зав. кафедрой патофизиологии Оренбургской государственной медицинской академии; Н.М. Беляева - кандидат мед. наук, доцент кафедры патофизиологии Оренбургской государственной медицинской академии.
Под общей редакцией д.м.н., проф. Б.А. Фролова.
Рецензенты: Зав. кафедрой патофизиологии ГОУ ВПО "Челябинская государственная медицинская академия Росздрава" доктор мед. наук, профессор Л.В. Кривохижина; Зав. кафедрой патофизиологии Башкирского государственного медицинского университета, засл. деятель науки РБ, акад. РАЕН, доктор мед. наук, профессор Д.А. Еникеев; Профессор кафедры клеточной биологии и гистологии биологического факультета МГУ им. М.В. Ломоносова доктор биол. наук. Г.Е. Онищенко; Профессор кафедры онкологии I ММА им. И.М. Сеченова доктор мед. наук Д.Ш. Османов.
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
УЧЕБНО-МЕТОДИЧЕСКОЕ ОБЪЕДИНЕНИЕ ПО МЕДИЦИНСКОМУ И ФАРМАЦЕВТИЧЕСКОМУ ОБРАЗОВАНИЮ ВУЗОВ РОССИИ
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ ______________ МОСКОВСКАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ им.И.М.СЕЧЕНОВА ______________ Москва М.Трубецкая, д. 8, корп.2 Тел. 248-53-21,246-24-11,247-11-80(факс)
УМО - № 149- Д 28.03.06 Ректору Оренбургской государственной медицинской академии, профессору С.А.Павловичеву
Учебно-методическое объединение по медицинскому и фармацевтическому образованию вузов России, рассмотрев материалы рукописи:
Б.А.Фролов, Н.М.Беляева «Молекулярные механизмы опухолевого роста»
и сообщает, что указанным рукописям присвоен гриф УМО:
"Рекомендуется Учебно-методическим объединением по медицинскому и фармацевтическому образованию вузов России в качестве учебного пособия для студентов медицинских вузов".
Проректор ММА им. И.М.Сеченова, зам. председателя УМО медицинских и фармацевтических вузов, профессор
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
В последние десятилетия достигнут значительный прогресс в выяснении молекулярных механизмов канцерогенеза. Идентифицированы генетические дефекты, определяющие необходимые и достаточные свойства раковой клетки: постоянную митогенную стимуляцию, нечувствительность к антиростовым и проапоптотическим сигналам, неограниченный пролиферативный потенциал, способность индуцировать ангиогенез, инвазию и метастазирование. Сформулировано представление о многостадийности (многоступенчатости) канцерогенеза, как процесса накопления в клетке генетических дефектов, вызывающих дискретные и необратимые изменения ее генотипа. Выявлены функциональные связи опухоли с нормальным окружением и установлена поразительная способность раковых клеток рекрутировать окружающие здоровые ткани для стимуляции собственного роста и прогрессии. Определены механизмы злокачественной инвазии и метастазирования и уточнена роль в этих процессах адгезивных взаимодействий опухолевых клеток друг с другом и с внеклеточным матриксом. Совершенствуются методы диагностики опухолей, основанные на определении опухолевых антигенов и онкофетальных белков. Разрабатываются принципиально новые молекулярно-биохимические подходы к терапии рака. Вышеизложенное диктует настоятельную необходимость обобщения современных представлений об опухолевом росте и его молекулярных механизмах, позволяющего помочь студенту разобраться в основных проблемах канцерогенеза, понять смысл и следствие новых событий в онкологии, а главное – подготовить его к восприятию новых научных идей. Мы полагаем, что решению этой задачи поможет настоящее пособие, содержащее иллюстративный материал, сопровождаемый лаконичными пояснениями. Знакомство с пособием, несомненно, предполагает достаточную подготовленность читателю по ряду вопросов в области биохимии, генетики, нормальной и патологической физиологии, цитологии – положение, наглядно демонстрирующее необходимость интеграции знаний по важнейшим медико-биологическим дисциплинам. Пособие снабжено тестовыми заданиями и вопросами, обеспечивающими возможность самоконтроля знаний.
Раздел I. Раздел II.
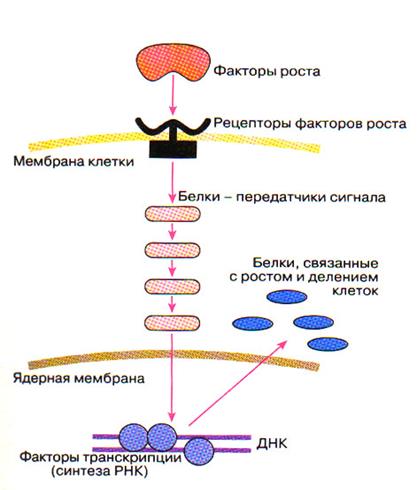
МЕХАНИЗМЫ ГЕНЕТИЧЕСКОГО КОНТРОЛЯ КЛЕТОЧНОГО ЦИКЛА. ИНДУКЦИЯ КЛЕТОЧНОГО ЦИКЛА Основные этапы передачи сигнала для роста от мембраны до клеточного ядра (Г.П. Георгиев, 1999) · Клетки организма находятся в одном из трех возможных состояний: в цикле, в стадии покоя с сохранением возможности вернуться в цикл и в стадии терминальной дифференцировки, при которой способность делиться полностью утрачена (нейроны головного мозга, сегментоядерные нейтрофилы). · В норме стимуляция клеточного роста (вхождение клетки в цикл) инициируется факторами роста, которые воспринимаются с помощью соответствующих рецепторов. · Последующая трансдукция сигнала от активированного рецептора до клеточного ядра осуществляется с участием многих других белков - передатчиков сигнала. Это своего рода клеточное "реле". Такая передача часто идет путем фосфорилирования одним белком второго, вторым - третьего и т.д. · Цепи передачи сигналов заканчиваются в клеточном ядре. Там происходит активация так называемых факторов транскрипции, т.е. белков, связывающихся с регуляторными участками определенных генов ДНК и активирующих транскрипцию данных генов. Иными словами, под действием факторов транскрипции на соответствующих генах происходит синтез матричных РНК, а на матрице последних - белков. Это те белки, которые нужны для роста и размножения клеток. Раздел III. ОПРЕДЕЛЕНИЕ ПОНЯТИЙ (по Б.П. Копнину, 2000; А.В. Лихтенштейну, В.С. Шапоту, 2001) · Онкогены - гены (последовательности нуклеиновых кислот), обусловливающие неконтролируемый опухолевый рост клеток (in vivo) и их трансформацию в культуре (in vitro). Выделяют вирусные (v-onc) и клеточные (c-onc) онкогены.
· Вирусные онкогены - трансформирующие гены в составе высокоонкогенных РНК и ДНК -содержащих вирусов.
· Онкогены ДНК-содержащих опухолеродных вирусов вирусспецифичны и клеточных аналогов не имеют.
· Онкогены РНК-содержащих опухолеродных вирусов (ретровирусов) - исходно не вирусного происхождения и не нужны для размножения вирусов. Они происходят из протоонкогенов, захваченных вирусом из генома позвоночных во время инфекционного цикла.
· Протоонкогены - клеточные гомологи вирусных (ретровирусы) онкогенов в геноме позвоночных. Это - нормальные гены, контролирующие синтез факторов роста, белков-рецепторов, белков-трансдукторов митогенного сигнала, факторов транскрипции, а через них - процессы клеточной пролиферации, дифференцировки и морфогенеза. Протоонкогены - элементы позитивной регуляции клеточного деления. Они являются его акселераторами.
· Не все протоонкогены, обнаруженные в геноме человека, имеют вирусные аналоги.
· В нормальной клетке протоонкогены характеризуются адекватным функционированием в соответствии с поступающими сигналами, определяющими потребность клетки в контролируемых ими белках. Если протоонкогены переходят в перманентно активное состояние вне зависимости от поступающих сигналов, они вызывают неконтролируемый рост и трансформацию клетки. В этом случае их называют клеточными онкогенами (с-onc).
· Активированные с-onc независимы от клеточного генома. Они инициируют и поддерживают размножение трансформированных клеток, активируя сигнальные механизмы митотического цикла. · Структура онкогенов ретровирусов отличается от структуры протоонкогенов. Большинство из этих структурных изменений обусловливает возможность v-onc встраиваться в хромосомы хозяина и индуцировать активацию клеточных протоонкогенов, превращая их в c-onc. ОПРЕДЕЛЕНИЕ ПОНЯТИЙ (продолжение)
· Протоонкогены, в том числе гомологичные вирусным онкогенам, могут активироваться и без участия вирусов в результате процессов хромосомных транслокаций, амплификаций, мутаций.
· В нормальных клетках присутствуют белковые факторы негативной регуляции клеточного деления. Кодирующие их гены носят название генов-супрессоров (Rb, р53, АРС и др.). Реализация эффектов онкогенов находится в прямой зависимости от супрессии механизмов негативной регуляции клеточного деления.
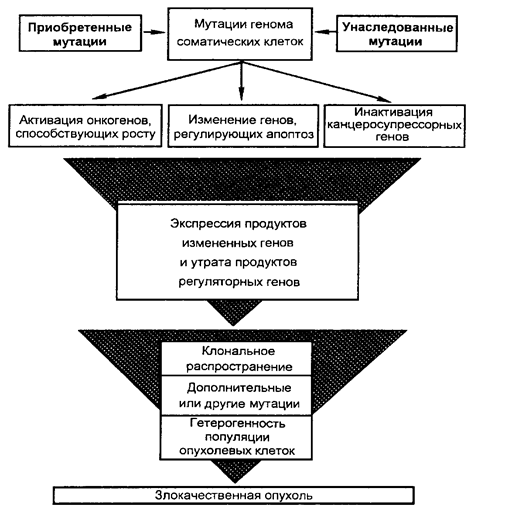
· Таким образом, полная трасформация клетки является многоступенчатым процессом для достижения которого необходимо несколько генетических событий: активации онкогенов и инактивации генов, осуществляющих супрессорную функцию. При этом разные онкобелки действуют по общим биохимическим путям, в связи с чем разные онкогены могут дополнять эффекты друг друга, но один и тот же онкоген способен вызвать разные эффекты в клетках различного гистогенеза.
· Непрерывность роста трансформированных клеток поддерживается их нечувствительностью к апоптозу вследствие угнетения продукции проапоптотических факторов и усиления продукции антиапоптотических факторов:
Раздел IV. Раздел V. Общие положения Различают антиканцерогенные, антимутационные и антицеллюлярные механизмы противоопухолевой защиты (П.Ф. Литвицкий, 2002). Антиканцерогенные механизмы Антимутационные механизмы Антицеллюлярные механизмы СИСТЕМА РЕПАРАЦИИ ММR

Механизм регуляции MMR (поА.В. Лихтенштейну, В.С. Шапоту, 2001)
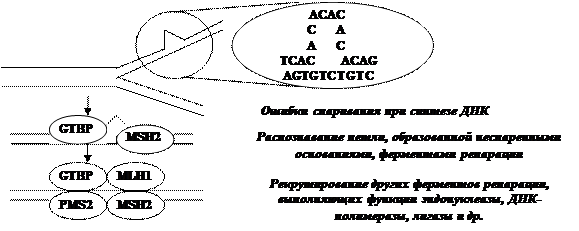
· Инициация и прогрессия опухоли - суть фенотипические проявления нарушений генотипа (ДНК). В связи с этим противоопухолевая защита клетки осуществляется не только через механизмы супрессии клеточной пролиферации и активации апоптоза, но и путем репарации ДНК - восстановления нарушенной структуры. · Нарушения структуры ДНК могут быть следствием не только модификации или повреждения оснований и нуклеотидов, что характерно для действия физических и химических мутагенов. Они могут быть и результатом ошибок спаривания оснований, при которых вместо комплементарной пары нуклеотидов А+Т или Г+Ц в дочернюю цепь оказываются включенными нуклеотиды, некомплементарные нуклеотидам в материнской цепи ДНК. Эти некомплементарные нуклеотиды называют мисмэтчами (mismatch). · Возникновение подобных нарушений структуры ДНК возможно из-за ошибочной подстановки нуклеотида ДНК-полимеразой, чему способствует наличие в геноме многократных повторов 2-3 нуклеотидов, образующих т.н. микросателлитные последовательности. В геноме человека присутствует около 100.000 повторов ЦА/ГТ. При репликации таких локусов и возможен эффект скольжения(slippage), что приводит к образованию неспаренных участков, которые затрагивают только дочернюю нить ДНК. · Удаление подобных дефектов структуры ДНК - мисмэтч - репарация - прерогатива системы MMR (mutations in mismatch repair), которую обеспечивают несколько согласованно действующих белков. Одни из них (МSH2, GТВР) распознают дефект связывания с участками неспаренности и рекрутируют другие белки (МLН1, РМS2). Последние восстанавливают структуру дочерней нити ДНК, полностью комплементарную материнской нити. · При дефектах ММR возникает т. н. мутаторный фенотип. Частота мутаций возрастает в 100-1000 раз. Нестабильность микросателлитов охватывает весь геном и отражается на функционировании многих генов, в составе или вблизи которых расположены микросателлиты. · Неполноценность ММR резко увеличивает вероятность новых мутаций, которые, лавинообразно накапливаясь в ряду клеточных поколений, способствуют ускоренной прогрессии опухоли. Дефекты МLH1 и МSН2 обнаружены при различных семейных и спорадических формах рака, в частности, при раке толстой кишки. · Поскольку гены ММR относятся к числу генов-супрессоров, мутаторный фенотип возникает при инактивации обеих аллелей соответствующего гена. ГЕН p53 И ЕГО РОЛЬ В ОБЕСПЕЧЕНИИ СТАБИЛЬНОСТИ ГЕНОМА ПРИ ПОВРЕЖДЕНИЯХ ДНК
· Сенсором повреждения ДНК и нарушения в клеточном цикле является ген р53. Этот ген, располагающийся на коротком плече хромосомы 17, кодирует образование ядерного белка из 393 аминокислот с молекулярной массой 53 кД. Тетрамер р53 локализован в клеточном ядре и функционирует как транскрипционный фактор, связываясь своим карбоксильным окончанием со специфическими регионами генов-мишеней. Показано, что активация гена р53 происходит не только в ответ на поражение ДНК (под действием ионизирующего или УФЛ-облучения, ингибитора топоизомеразы 2 и прочих факторов), но может явиться следствием многих других процессов, происходящих в клетке, в том числе активации онкогенов, гипоксии, дефиците питания, старении и др. Недаром этот ген получил название "хранитель генома" (Е.Б. Владимирская, 2002). · При активации гена р53, кодируемый им белок р53 способен инициировать независимо друг от друга две программы (Н.Н. Белушкина, С.Е. Северин, 2001): а) временную остановку клеточного цикла в G1/S-фазе с помощью белка р21WAF1, ингибирующего циклинзависимые киназы; б) стимуляцию апоптоза путем активации генов Bax или Bid - проапоптотических генов семейства Bcl 2 и/или активации образования свободных форм кислорода, способствующих выходу цитохрома С из митохондрий. · Блок клеточного цикла в фазе G1 - фазе репликации ДНК, делает возможной репарацию поврежденной ДНК и предотвращает тем самым появление мутантных клеток. Считают, что приоритетной для большинства клеток является программа временной остановки митотического цикла, хотя выбор стратегии может характеризоваться тканеспецифичностью. · Есть данные об участии р53 в процессах репарации ДНК путем активации вновь открытого гена р53R2, кодирующего рибонуклеотидредуктазу. · Программа апоптоза включается при невозможности клетки репарировать ДНК во время "ареста" при прохождении митотического цикла, или дефиците белка р21WAF1. · Т.о., основной функцией гена р53 и кодируемого им белка (р53) следует считать защиту организма от накопления генетически дефектных клеток. Мутация гена р53 позволяет таким клеткам сохранять жизнеспособность в митозе, что чревато их выживанием и, следовательно, развитием опухолевого процесса. Такие мутации связаны с плохим прогнозом в лечении злокачественных новообразований, поскольку опухолевые клетки с этими мутациями оказываются резистентными к лучевой и химиотерапии. · Мутация гена р53 обнаруживаются более, чем в половине раковых опухолей. При длительной химиотерапии частота ее повышается. У детей мутация в гене р53 чаще наблюдается при остром Т-лимфобластном лейкозе, составляя около 12% и всегда служит прогностически неблагоприятным фактором.
РЕЗЮМЕ
· В основе многоклеточности, которая служит фоном для развития опухоли, лежит способность каждой клетки интегрировать сигналы, поступающие от растворимых факторов, взаимодействий клеток с внеклеточным матриксом и друг с другом. Правильная обработка этих сигналов клеткой создает основы для нормального клеточного роста, дифференцировки и морфогенеза. Некорректная интеграция поступающих с разных сторон сигналов, приводит к развитию патологии, в частности, рака (Е.Д. Свердлов, 2001).
· Рак вызывается эволюцией клеток, выражающейся в последовательном накоплении не связанных между собой мутаций, делающих клетку все более автономной от окружения. Эти мутации затрагивают как онкогены (ras и др), способствующие пролиферации, так и гены-супрессоры, участвующие в контроле клеточного цикла (Rb) и обеспечивающие стабильность генома (р53, APC, BRCA, ATM и др.)
· Независимость мутаций отдельных генов не означает независимости действия в клетке каждого из контролируемых ими продуктов, которые выполняют функцию сигнальных молекул. Включаемые этими молекулами различные линейные сигнальные пути объединяются в общую сигнальную сеть (networks) и, таким образом, переплетаются. Т.е. онкогены и гены-супрессоры действуют зависимо друг от друга и при опухолевой трансформации изменяются не единицы и не десятки, а сотни генов. Таким образом, в раковую трансформацию вовлекаются не отдельные звенья регуляции генома, а интегральные процессы.
· Независимость опухоли от окружения не означает ее абсолютной изолированности: "нормальные клетки" внутри раковой опухоли оказываются активными участниками опухолевого процесса и используются раковыми клетками для своего существования. В частности, ростовые сигналы, управляющие пролиферацией клеток карциномы могут происходить от клеток нормальной стромы - компонентов опухолевой массы.
· "Успешными" опухолевыми клетками являются те, которые приобретают способность использовать своих нормальных соседей, индуцируя их испускать потоки сигналов роста для увеличения потенциала роста опухоли.
· В развивающейся опухоли идет непрерывное взаимодействие между системой передачи сигналов и изменениями генома. Изменения генома, улучшающие сигнализацию, могут приводить к дальнейшему ускорению изменения генома и т.д.
· Таким образом, опухоль превращается в сообщество множества типов клеток, которые коэволюционизируют, обмениваясь сигналами и изменяя структуру генома так, чтобы оптимизировать эволюцию опухоли, в том числе, возможно, путем оптимизации обмена сигналами между различными компонентами эволюционизирующей биомассы. Раздел VI. КАНЦЕРОГЕНЕЗ. ЭТИОЛОГИЧЕСКИЕ ФАКТОРЫ КАНЦЕРОГЕНЕЗА
· Рак - болезнь генов. Различные факторы, способные вызвать нарушения в геноме клетки (проявление генотоксичности) обладают канцерогенным действием, т.е. способностью вызывать или ускорять развитие новообразований. Они получили название канцерогенов. · По определению ВОЗ «Канцерогеном (физическим, химическим, вирусным) называют агент, способный вызывать или ускорять развитие новообразования независимо от механизма (или механизмов) его действия или степени специфичности его эффекта. Канцероген –это агент, который в силу своих физических или химических свойств может вызвать необратимое изменение или повреждение в тех частях генетического материала, которые осуществляют гомеостатический контроль над соматическими клетками». · В подавляющем большинстве случаев определить конкретный канцероген в качестве причины заболевания невозможно в силу по меньшей мере двух обстоятельств: - присутствия великого множества канцерогенов в окружающей и внутренней среде организма; - многостадийности канцерогенеза, при которой каждая последующая стадия может быть спровоцирована разными факторами. · Наряду с канцерогенами (как этиологическими факторами) выделяют факторы риска опухолевого роста, т.е. факторы, способные усилить (ускорить) проявления действия причины. К ним относятся - Возраст. Рак – болезнь пожилых людей. После 55 лет его вероятность прогрессивно нарастает. При старении и сопутствующих заболеваниях риск опухолевого роста обусловлен: 1) увеличением повреждаемости ДНК при снижении способности к репарции; 2) метаболическими сдвигами в виде усиления образования свободных радикалов; 3) эндокринным дисбалансом; 4) ослаблением иммунологического контроля. - Хронические пролиферативные заболевания в виде метаболических, дисгормональных и воспалительных процессов, оказывающих воздействие на генетический контроль над пролиферацией и дифференцировкой клеток и вызывающих факультативные предраковые изменения. - Стиль жизни. Наличие вредных привычек (курение, алкоголизм), особенности питания, традиции. - Первичные и вторичные иммунодефициты, ослабляющие механизмы распознавания и отторжения (уничтожения) опухолевых клеток. - Наследственность. Несмотря на генетическую природу рак не наследственное заболевание, поскольку лежащие в его основе соматические мутации не передаются по наследству. Лишь 7% случаев рака связаны наследственной предрасположенностью.
ХИМИЧЕСКИЙ КАНЦЕРОГЕНЕЗ Экзогенные химические канцерогенные вещества (по В. В. Худолею, 1999)
· Химические канцерогены подразделяются на проканцерогены (составляют абсолютное большинство) и прямые канцерогены.
· Проканцерогены превращаются в истинные, конечные канцерогены только после метаболических превращений, катализируемых тканевыми ферментами (неспецифическими оксидазами). Они локализованы, главным образом, в ЭПР и частично в ядре клетки. Так, ПАУ становятся конечными канцерогенами, превращаясь в соответствующие эпоксиды; нитрозамины – в диазоалканы.
· Некоторые проканцерогены становятся конечными канцерогенами в результате спонтанных реакций.
ХИМИЧЕСКИЙ КАНЦЕРОГЕНЕЗ ВИРУСНЫЙ КАНЦЕРОГЕНЕЗ ВИРУСНЫЙ КАНЦЕРОГЕНЕЗ ВИРУСНЫЙ КАНЦЕРОГЕНЕЗ ВИРУСНЫЙ КАНЦЕРОГЕНЕЗ КАНЦЕРОГЕНЕЗ. ПРОМОЦИЯ · Промоция – стадия реализация опухолевого фенотипа обусловлена влиянием различных факторов – промоторов, не обладающих способностью вызывать повреждения ДНК, не являющихся канцерогенами. Действие промоторов на инициированные клетки стимулирует их деление, обеспечивая наработку «критической массы» опухолевых клеток, повышающей их устойчивость и автономность роста. Ряд промоторов - форболовые эфиры (TPA) и их производные - агонисты протеинкиназы С (ПКС). При участии РКС осуществляется мобилизация многих внутриклеточных систем, позволяющих клетке избежать апоптоз и гибель от апоптоза и различных неблагоприятных воздействий. · В отличие от инициаторов действие промоторов не затрагивает ДНК и поэтому обратимо. Для них существует пороговый уровень действия, поэтому субпороговые или раздельные дозы с большими паузами между ними не дают завершающего канцерогенного эффекта.
Патогенез злокачественных новообразований (М.А. Пальцев, Н.М. Аничков, 2001) · Канцерогенез с раздельным участием инициаторов и промоторов реализуется при действии неполных канцерогенов, канцерогенов в низких дозах (ДМБА) или некоторых онкогенных вирусов (Эпштейн-Барра). · Полные канцерогены (нитрозамины), ионизирующее излучение сами обладают как нициирующим, так и промоцирующим действием. Общие положения
Опухолевая прогрессия, гетерогенность опухоли (по V. Cumar, R. S. Cotran, S.L. Robbins, 1997)
· «Опухолевая прогрессия» (Л.Фулдс, 1969) - генетически закрепленные, наследуемые опухолевой клеткой и необратимые изменения одного или нескольких ее свойств. · Основа опухолевой прогрессии – нестабильность генома, обусловленная множественными мутациями. · Изменения различных свойств клеток бластомы происходят независимо друг от друга, поскольку мутации каждого гена автономны (правило независимости опухолевой прогрессии). · Фенотипически опухолевая прогрессия проявляется изменением биохимических, морфологических, электрофизиологических, антигенных и функциональных признаков опухоли. · Сроки изменений свойств разных клеток бластомы сильно варьируют. В связи с этим признаки их появляются и изменяются без какой либо закономерной хронологии. · При опухолевой прогрессии создаются субклоны клеток с самой различной комбинацией признаков. При этом разные субклоны клеток одного и того же новообразования могут существенно отличаться друг от друга. · Результатом прогрессии является приобретение селективного преимущества субклонов опухолевых клеток, обладающих наибольшей резистентностью к действию защитных механизмов и наибольшей агрессивностью, т.е. способностью к инвазии и метастазированию (клональная селекция). Раздел VIII. Раздел IХ. Раздел Х. ЕСТЕСТВЕННЫЕ КИЛЛЕРЫ (А.А. Ярилин, 1999; Р.М. Хаитов с соавт., 2000) · Для реализации активности NK не требуется их предварительного контакта с индуцирующим агентом и развития иммунного ответа. Они лишены антигенраспознающих рецепторов. В фило- и онтогенезе система естественных киллеров появляется раньше, чем факторы антигенспецифической иммунной защиты. Данная система ответственна за противоопухолевую и противовирусную резистентность организма. · В отличие от Т-лимфоцитов, NK-лимфоциты убивают клетки-мишени быстро (1-2 часа), используя упрощенный механизм без распознавания индивидуальных антигенов и подготовки в виде иммунного ответа. · Рецептор NK-клеток, предназначенный для распознавания клеток-мишеней представляет собой С-лектин, т.е. белок, распознающий углеводные остатки при участии ионов Са2+. Этот рецептор - NK R - Р1 (CD 161) распознает концевые остатки маннозы на молекулах мембранных гликопротеинов и гликолипидов. В норме такие остатки на большинстве клеток, с которыми контактируют зрелые лимфоциты и макрофаги, блокированы молекулами сиаловой кислоты. Это защищает их от фагоцитоза макрофагами (которые также имеют рецепторы, связывающие маннозу) и от лизиса NK. · Свободная манноза присутствует на поверхности юных и старых клеток, а также на пролиферирующих и трансформированных клетках. · Способность убивать клетки опухолевых линий у NK ограничена. Механизм этого ограничения связан с возможностью распознавания последними аутологичных молекул MHC при участии соответствующих рецепторов (KIR: NKB-1). В случае такого распознавания в NK-клетку передается сигнал, запрещающий развитие дальнейших событий, ведущих к цитолизу. В результате мишенями NK могут быть клетки, на поверхности которых присутствуют глюкоконъюгаты со свободными остатками маннозы и не содержатся молекулы МНС I класса (мера, предупреждающая повреждение собственных клеток организма). · При соблюдении этих условий происходит сближение NK-клеток с их мишенями, которое определяется не только связыванием рецепторов к глюкоконъюгатам, но и адгезивными взаимодействиями, в реализации которых принимают участие ганглиозиды, интегрины и другие молекулы. · После установления подобного контакта запускается программа апоптоза клетки-мишени с участием перфорин-зависимого механизма. · NK-клетки способны осуществлять рециклинг, т.е. отсоединяться от погибшей клетки и участвовать в повторном киллинге новых клеток-мишеней. ПЕРФОРИН-ЗАВИСИМЫЙ АПОПТОЗ
ЦИТОТОКСИЧЕСКИЕ Т-ЛИМФОЦИТЫ (Р.М. Хаитов с соавт., 2000) · Антигенспецифическая цитотоксичность обеспечивается дифференцированной субпопуляцией CD8+ Т α β - лимфоцитов: ЦТЛ. Рецептор этих лимфоцитов распознает антиген в комплексе с молекулами MHC-1 на мембране клеток-мишеней, что является необходимым условием для киллерной атаки ЦТЛ. · Главное защитное предназначение ЦТЛ - санация организма от внутриклеточных инфекций. · Неиммунные зрелые ЦТЛ 8+ имеют только программу для биосинтеза эффекторных молекул (цитотоксинов), которая начинает реализовываться после вовлечения их в иммунный ответ. Для этого лимфоциту недостаточно только связать распознаваемый антиген в комплексе с молекулой МНС на той или иной клетке. Требуются также и молекулы костимуляции. Если опухоль растет не из профессиональных антигенпредставляющих клеток, то этих молекул костимуляции на опухолевых клетках нет. И за фактом распознавания лимфоцитом (в тех случаях, когда есть что распознавать) не следует иммунный ответ, пока профессиональные антигенпредставляющие клетки не процессируют опухольспецифичные антигены. Обычно условия для этого отсутствуют. · В том случае, когда ЦТЛ 8+ вовлекаются в иммунный ответ в них происходит синтез de novo определенных веществ, которые в виде функционально неактивных молекул-предшественников накапливаются в гранулах. Эти гранулы сориентированы в связи с TCR, чем обеспечивается возможность строго локального киллерного удара по клетки-мишени. · Цитотоксины гранул ЦТЛ как минимум представлены двумя белками: перфорином и гранзимами (сериновыми протеазами), организующими сигнал на апоптоз для клетки-мишени. · Собственно механизм киллинга при участии ЦТЛ состоит в том, что связывая своим TCR антиген на поверхности клетки-мишени, ЦТЛ в области этой связи быстро формируют межклеточный интерфейс - локальную зону контакта с последующим выбрасыванием содержимого гранул. · В отличие от NK, ЦТЛ обладают еще |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-07-15; просмотров: 835; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.141 (0.012 с.) |

 И.Н.Денисов
И.Н.Денисов