
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Параметри системи при змішуванні розчинівСодержание книги
Поиск на нашем сайте
х – умовна доля води, яка залежить від співвідношення
Теплота кристалізації Qк дорівнює залишковій диференціальній теплоті розчинення L, але оборотна їй за знаком. Теплоту кристалізації (в кДж/моль) можна розраховувати користуючись номограмою (рис.2.6). Наприклад, маючи дані про розчинність речовини при двох температурах, з'єднують на монограмі значення концентрації і температури прямими лініями. Нехай розчинність першої речовини при 20° С дорівнює 20%, а другої при температурі 60° С – 50 %.
На лівій ординаті відмічаємо точку lnC1 = ln20 =3,86 і з'єднуємо її з точкою t1 = 30° C на правій ординаті. Потім точку lnC2 ln50 = 5,28 з'єднуємо з точкою t2 = 60°C. З фігуративної точки f перетину цих двох прямих опускаємо перпендикуляр на вісь абсцис і визначаємо значення ln Qk, яке дорівнює 2,6. Звідси Qk = 13,4 кДж /моль. Значення теплот кристалізації, які знаходять за номограмою або розраховують теоретично не є точними, тому більш надійною величиною є теплота розчинення, що визначається експериментально. Знак величини теплоти кристалізації речовин залежить від їх розчинності. Якщо для речовини температурний коефіцієнт розчинності:
то теплоту кристалізації необхідно відводити, а якщо
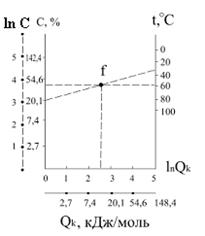
то для проведення процесу кристалізації необхідно підводити тепло. Теплота кристалізації кристалогідратів зростає зі зростанням кількості молекул води, що входять в склад кристалогідратів. При цьому екзотермічність перших молекул води кристалогідратів значно вище наступних молекул, внаслідок їх більш міцного утримання зі сполукою. Теплота розчинення, яку визначають експериментально, в загальному випадку складається із декількох складових: ΔНm =bDН ± +(1-b)DННЕДИС.+DНРОЗП.+DНРЕШ., (2.15) де β - степінь дисоціації при концентрації Сm; ΔН± - сумарна теплота сольватації (гідратації) катіона і аніона; ΔННЕДИС. - теплота утворення недисоційованної частини їз йонів речовини, що умовно знаходяться в газоподібному стані, і розчинника; ΔНРОЗП. - теплота розподілення розчиненої речовини по об'єму розчину; ΔНРЕШ. - теплота руйнування кристалічної решітки сполуки, що розчиняється. Ймовірно, що ΔНРЕШ. характерна тільки для конденсованих твердих речовин. Перші три складових рівняння характеризують сольваційно-дисоційні теплові ефекти, до яких можуть бути добавлені теплоти інших фізико-хімічних взаємодій. Насичені і пересичені розчини. Насичений розчин характеризується стійкою рівновагою розчину з надлишком розчиненої речовини при даній температурі. Стан насичення розчину встановлюється при рівності:
де mі, mіо - хімічні потенціали йонної речовини, що перебуває відповідно в фазі, яка розчиняється, і в насиченому розчині. Отже, насиченим розчином називається розчин, що містить максимальну кількість речовини, яка може розчинитися в даній кількості розчинника за даної температури. Розчини, що містять більшу або меншу кількість розчиненої речовини називаються відповідно пересиченими і ненасиченими. Утворення пересиченого розчину можливе під час повільного охолодження розчину, в якому є утруднення з виникненням зародків нової фази (центрів кристалізації), і система переходить в метастабільний стан – пересичений розчин. Цей перехід відповідає потенціалу переносу маси Dm речовини, що розчиняється в насичений розчин, який можна виразити як:
Стан пересичення бінарних розчинів можна проілюструвати рис.2.7, на якому видно розташування областей ненасиченого (І),
пересиченого метастабільного (ІІ) і лабільного (ІІІ) розчинів, ліній 1,1’, які відповідають концентраціям насичених розчинів, ліній 2,2’ – ліній розділення метастабільного і лабільного пересичених станів. На рисунку 2.7 виділені політерми підвищення розчинності і зниження розчинності (точки з індексами „штрих”) з підвищенням температури. Шляхом ізотермічного випаровування частини розчинника (лінія NK) в системі утворюється пересичення: ∆CВИП. = СK – СN. Охолодженням насиченого розчину (точка N,CN) від температури ∆CОХ. = СN - СL. У випадку зниження розчинності з підвищенням температури (політерма 1ˈ) шляхом випаровування розчинника із розчину, насиченого при температурі
Нагрівання насиченого розчину від
тобто стан пересичення розчинів можна характеризувати температурними перепадами ∆Т, які відповідають собою переохолодженню або перегріву системи. Явище пересичення можливе і під час хімічної взаємодії, що відбувається між компонентами насиченого розчину. Наприклад, в насиченому розчині СаСО3 (5 мг/дм3) йони Са2+ можуть зв'язуватись йонами Пересичена система, що знаходиться в ізотермічних умовах і ізольована від впливу фізичних і хімічних факторів, тривалий час може бути досить стійкою. В неї важкорозчинна сполука AmBn, що дисоціює за реакцією: AmBn L mAn+ + nBm-, для якої
і йонний добуток В насиченому розчині, в якому Розчинність осадів залежить від величини ДР осаду, а також від надлишку добавленого йону, що входить в склад осаду:
Добавлення сторонніх електролітів, що не мають спільних йонів з речовиною, яка випала в осад, спричинює збільшенню йонної сили розчину внаслідок чого розчинність осаду збільшується (сольовий ефект). Так, наприклад у присутності 1 моль/л KNO3 розчинність PbSO4 збільшується в 14 разів, а в присутності 1 моль/л Al(NO3)3 – в 77 разів. Це пояснюється тим, що „сторонні” йони утримують йони „хазяїв” розчину у розчиненому стані, який перевищує розчинність малорозчинної солі в чистому розчиннику. Розчинність важкорозчинних солей залежить від концентрації йонів гідрогену в розчині:
де КД – константа дисоціації кислоти [H+] = CHB-[HB ] – концентрація йонів гідрогену, що дорівнює різниці між загальною концентрацією кислоти і концентрацією її недисоційованої частини. Залежність розчинності електролітів від йонної сили розчину. Основи теорії сильних електролітів, які розроблені П.Дебаем і Е.Хюккелем, придатні лише для розбавлених розчинів сильних електролітів до концентрації не вище ніж 1 . 10-3 моль/л. За теорією кожний центральний йон оточений йонною сферою, що складається з негативно і позитивно заряджених йонів. Сумарний заряд йонів і йонної сфери дорівнює за величиною і протилежний за знаком заряду центрального йону. Потенціал йонної сфери
де: zi – заряд йону, е – заряд електрону; Д – відносна діелектрична проникність розчинника;
де сі – концентрація йону; NA – число Авогадро; k – константа Больцмана, Т – температура. Для врахування відхилення властивостей концентрованих розчинів електролітів від теорії електролітичної дисоціації вводять поправку на ідеальність розчину
де Коефіцієнт активності пов’язаний з активністю речовини аі:
Мірою електричної взаємодії між йонами в розчині є йонна сила І:
яка залежить від концентрації і зарядів йонів, що знаходяться в розчині. У загальному випадку йонну силу розчину електроліту, що містить катіони і аніони визначають за виразом:
де [Kt], [An] – концентрації відповідно катіонів і аніонів, моль/л; Залежність коефіцієнта активності йону від йонної сили розбавленого розчину виражається рівнянням Дебая-Хюккеля:
а у випадку водних розчинів сильних одно-одновалентних електролітів з концентрацією до 1 . 10-3 моль/л за рівнянням:
де се – концентрація електроліту. Йонна сила для розчинів одно-одновалентних електролітів (
для одно-двовалентних (
для дво-двовалентних (
а для одно-тривалентних (К3РО4) і три-одновалентних (А1С13):
Якщо в розчині знаходиться суміш електролітів, то враховують сумарні концентрації кожного виду катіонів і аніонів, наприклад, йонна сила 0,001 М розчину
а коефіцієнт активності:
Активність йонів буде:
У випадку змішування розчинів, наприклад KNO3 і K2SO4 в суміші якого міститься 0,02 моль/л КС1 і 0,08 моль/л K2SO4 сумарна концентрація йонів калію становить
Тоді йонна сила розчину суміші солей буде:
Внаслідок сильної гідратації катіонів і слабкої гідратації великих полівалентних аніонів електроліти з багатовалентними катіонами мають вищі коефіцієнти активності ніж аналогічні електроліти з полівалентними аніонами:
З однаковими аніонами коефіцієнти активності електролітів зменшуються в міру збільшення заряду катіона:
Коефіцієнти активності електролітів, які утворені одновалентними катіоном і аніоном, зменшуються в такому ряду:
якщо електроліти утворені одновалентними катіонами У випадку однакових катіонів коефіцієнти активності знижуються в міру збільшення заряду аніона:
З одно-одновалентних електролітів з однаковими аніонами найбільші коефіцієнти активності мають кислоти, а коефіцієнт активності гідроксидів змінюється в зворотному порядку ніж одновалентних катіонів. Слід відмітити, що високі коефіцієнти активності мають хлорати (VII) двовалентних металів. Зі зростанням концентрації розбавлених розчинів електролітів коефіцієнт активності зменшується. При цьому крива залежності в багатьох випадках має мінімум, яка при подальшому підвищенні концентрації зростає і може набувати великих значень. У сильно розбавленому розчині, важкорозчинного електроліту, коли:
процес розчинення вже не залежить від йонної сили розчину, при добавленні до нього добре розчинного сильного електроліту відбувається збільшення не тільки йонної сили розчину, а і розчинності важкорозчинного електроліту. Це пояснює механізм сольового ефекту, розглянутого раніше. Для одно-одновалентного електроліту в цьому випадку добуток розчинності виражається як:
де Йонно-молекулярні рівняння в розчинах електролітів. Вивчення рівноважних йонно-молекулярних рівноваг в останній час актуально в агрохімії для характеристики степеню рухливості йонів ґрунтового розчину і в особливості грунтових фосфатів. Грунт представляє собою складну багатофазну систему, тому мінеральний фосфор в грунтовому розчині бере участь в багатьох рівноважних реакціях. Це реакції, які відбуваються тільки між рідкими фазами або тільки між твердими фазами, - гомогенні рівноваги: РРОЗЧИН L РРОЗЧИН - реакції, що відбуваються з великими швидкостями, РТВ.ФАЗА L РТВ.ФАЗА - реакції, що відбуваються з малими швидкостями. Реакції, що відбуваються між твердими і рідкими фазами – гетерогенні рівноваги: РГРУНТ L РРОЗЧИН. В результаті гомогенних реакцій ортофосфатна кислота і відповідні її йони Гетерогенні реакції включають розчинність і осадження важкорозчинних фосфорних солей, які характеризуються добутком розчинності цих сполук і адсорбцією фосфору на поверхні грунтових частинок. При внесенні в грунт фосфатних добрив, наприклад, гідрогенфосфату амоніаку, що містить вільну ортофосфатну кислоту під час розчинення солі відбувається вивільнення йонів за рахунок гідролізу катіонів слабкої основи: NH4+ + H2O L NH4OH + OH- та аніонів ортофосфатної кислоти за реакціями: PO43- + H2O L HPO42- + OH- ; HPO42- + H2O L H2PO42- + OH- ; H2PO4- + H2O L H3PO4 + OH- . Відповідно до констант йонізації гідроксиду амоніаку та ортофосфатної кислоти в грунтовому розчині встановлюються такі рівноваги: NH4OH L NH4+ + OH-; H3PO4 L H+ + H2PO4-; H2PO4- L H+ + H2PO42-; HPO42- L H+ + PO43-. Вміст окремих фосфат-йонів в грунтовому розчині можна розглядати, як функцію величини [H+] (рис.2.8).
Для кількісного розрахунку йонного складу розчину скористуємося рівняннями констант йонізації наведених сполук:
для ортофосфатної кислоти:
На стан рівноваги в розчині впливає рівновага дисоціації води, що описується відомою константою
Виходячи з умов електронейтральності для грунтового розчину, який розглядається, маємо рівновагу:
Кількість Нітрогену, Фосфору і Гідрогену в розчині можна розрахувати за рівнянням:
Враховуючи рівняння констант йонізації ортофосфатної кислоти і після перетворень маємо:
Застосувавши рівняння констант дисоціації гідроксиду амоніаку і води, концентрацію нітрогенвмісних речовин можна виразити через концентрацію йонів Гідрогену:
Таким чином, на підставі отриманих залежностей і рівняння матеріального балансу за йоном гідрогену після перетворень маємо тільки одну невідому величину:
Це рівняння розв’язують методами обчислювальної математики на ЕЦОМ. З графічної залежності (рис.2.8) випливає, що в грунтовому розчині більшості грунтів (рН = 3,5 - 8,5) присутні головним чином йони
Концентрації ортофосфатної кислоти виражають через активності відповідних йонів, тоді рівняння дисоціації кислоти приймають вигляд:
де Підставляючи
Після перетворень значення активності монофосфатного йону дорівнює:
Для дифосфатного йону аналогічно маємо:
де Гетерогенні реакції в грунті відбуваються, наприклад, при вапнуванні сильно кислих грунтів. При наявності в грунті вільної ортофосфатної кислоти в її розчині відбувається розчинення карбонату кальцію. В процесі розчинення карбонат кальцію дисоціює за рівнянням: CаCO3 L Са2+ + СO32-. При постійній температурі умова дисоціації характеризується добутком розчинності і розчинністю (S) карбонату кальцію:
В рівноважній гетерогенній системі необхідно враховувати інші рівноваги:
а також рівноваги ортофосфатної кислоти (див.вище). За умовою матеріального балансу розчинність карбонату кальцію дорівнює:
Перетворюючи ці рівняння рівняння матеріального балансу розчинності карбонату кальцію набуває вигляду:
Рівняння умови електронейтральності рівноважного розчину з урахуванням впливу ортофосфатної кислоти має вигляд:
Підставляючи вирази розчинності
рівняння умови електронейтральності набуває вигляду:
При цьому концентрацією гідроксид-йонів [OH-] в умовах кислого середовища знехтуємо. Рівноважні концентрації карбонатвмісних сполук можна записати, скориставшись виразами констант рівноваги карбонатної кислоти і умовою матеріального балансу розчинення карбонату кальцію, за допомогою рівноважної концентрації йонів гідрогену і констант йонізації:
Рівняння умови електронейтральності з урахуванням рівнянь констант йонізації Н3РО4, в яких Н3РО4=СР, а
Розв’язування цього рівняння дає можливість визначити рівноважну концентрацію [H+]. Запитання для самоконтролю 1. Чи залежить величина радіуса йонної атмосфери від концентрації йонів, їх природи, природи розчинника? Зробіть оцінку радіуса йонної атмосфери для 0,001 і 0,01 молярних розчинів 1,1- заряджених електролітів у воді згідно 1 наближенню теорії Дебая-Гюккеля. 2. Як відрізняють значення коефіцієнтів активності водних розчинів: а) КС1 і NaC12, б) КС1 і ВаС12, в) ВаС12 і BaSO4, якщо розчини однакової йонної сили (І випадок) і однакової молярної концентрації (ІІ випадок)? 3. Чому значення коефіцієнтів активності розчинів: КС1 у воді і КС1 в метиловому спирті відрізняються між собою? Відповідь підтвердити розрахунком. 4. Для 0,001 молярного водного розчину електролітів: NaCl, BaCl2, Na2SO4, ZnSO4 розрахувати значення коефіцієнтів активності 5. Обчислити коефіцієнти активності та активності йонів 0,01 М розчину Al2(SO4)3. 6. Як зміниться коефіцієнт активності 0,01 молярного водного розчину NaCl, якщо до нього добавили 0,015 моль/л Fe2(SO4)3 (T = 298 К)? 7. Визначити концентрацію йонів гідрогену і рН 0,5 %-го розчину сульфатної кислоти. 8. Як зміниться розчинність AgBrO3 в 0,02 молярному водному розчині AgNO3 порівняно з його розчинністю у воді (S = 0,0018 моль/л)?
Тема 3. Хімічна кінетика
Класифікація хімічних реакцій. Усі хімічні реакції проходять або в гомогенних системах, що складаються лише з однієї фази, або в гетерогенних, які складаються з двох і більше фаз. Такі реакції називаються відповідно гомогенними і гетерогенними. В гетерогенних системах одна із фаз завжди перебуває в диспергованому стані, тому хімічна взаємодія в них відбувається на поверхні поділу фаз. Розрізняють двофазні системи „рідина-тверда фаза”, „рідина-рідина” (що взаємонерозчинні), „газ-рідина”, “газ-тверда речовина”, „тверда речовина – тверда речовина”. Гетерогенні реакції відбуваються досить повільно ніж гомогенні. Це обумовлено складністю їх механізму: дифузія до поверхні поділу фаз, хімічна взаємодія реагентів, дифузія продуктів реакції від поверхні поділу фаз, поверхневі явища та ін. За механізмом усі реакції можна поділити на дві групи: прості і складні. Прості реакції складаються із однієї стадії – хімічного перетворення. За ознакою молекулярності вони поділяються на одномолекулярні (мономолекулярні), двомолекулярні (бімолекулярні), тримолекулярні. До складних реакцій відносяться ланцюгові, спряжені, паралельні, послідовні, оборотні, фотохімічні радіаційно-хімічні, ферментативні, гомогенно-гетерогенні. Для складних реакцій молекулярність можна визначити тільки для їх окремих стадій. Усі хімічні реакції формально поділяють на реакції першого, другого і третього порядків, а також нульового і дробного порядків. Під порядком реакції розуміють суму показників ступенів при змінюючих концентраціях реагентів в кінетичному рівнянні. Суму стехіометричних коефіцієнтів вихідних речовин, що одночасно беруть участь в елементарному акті реакції, прийнято визначати як її молекулярність. На швидкість гомогенних гетерогенних реакцій впливають: природа реагуючих речовин, концентрація речовин, тиск, температура, наявність каталізатора (ферментів). У випадку гетерогенних реакцій на їх швидкість також впливає ступінь диспергованості (поверхня поділу фаз) речовин. Знаючи хімічну кінетику реакцій можна визначати оптимальні параметри і умови проведення різних хіміко-технологічних і біохімічних процесів, які відбуваються при виробництві хімічних матеріалів, синтезу біологічних речовин, нейтралізації і знешкодження шкідливих речовин, забруднювачів навколишнього середовища. Швидкість хімічної реакції. Кінетичні закономірності перебігу хімічних реакцій ґрунтуються на припущенні про те, що реагують тільки ті молекули, які стикаються між собою. Кількість таких зіткнень прямо пропорційна кількості молекул, тому швидкість реакції повинна бути пропорційною концентрації реагуючих речовин (закон діючих мас). Для емпіричного рівняння:
Швидкість прямої реакції можна записати як:
де v - швидкість хімічної реакції; З рівняння випливає, що швидкість реакції, будучи функцією концентрації, залежить також від часу, оскільки концентрація реагуючих речовин з часом змінюється.
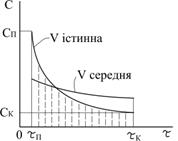
На рисунку 3.1 приведені графічні залежності зміни концентрації вихідного реагенту (крива 1) і продукту реакції (крива 2) з часом, а також середня і істина швидкості хімічної реакції.
Для заданого інтервалу часу
де За умови, що
швидкість хімічної реакції в даний момент часу відповідає значенню миттєвої (істинної) швидкості реакції v:
Для реагенту А середня швидкість реакції виразиться як:
В той же час, для продукту реакції D швидкість реакції буде мати знак плюс:
тому, що його концентрація збільшується. Швидкість реакції в даний момент часу можна визначити графічним способом. Для цього до точки на кривій зміни концентрації реагенту А, яка відповідає даному моменту часу (приклад Отже, хімічна кінетика визначає поняття швидкості гомогенної реакції як зміну концентрації одного з реагуючих компонентів за одиницю часу в одиниці об’єму (при постійній температурі):
де Гетерогенні реакції відбуваються на поверхні
У рівнянні швидкості прямої реакції коефіцієнт пропорційності
За цих умов:
Це і є фізичний зміст коефіцієнта На константу швидкості впливає в основному температура. Фактор впливу температури на константу швидкості гомогенної реакції виражає рівняння Арреніуса:
де А і В – характерні для даної реакції сталі, що не залежать від температури.
Рівняння Арреніуса в диференціальній формі має вигляд:
Величина Е називається арреніусовською енергією активації. Її розглядають як енергетичний бар’єр, що подолають лише активні молекули. Значення енергії активації дістають з експериментальних даних. Для цього будують графічну залежність lnk і 1/T в прямокутній системі координат. Ця залежність є прямою лінією, за кутом нахилу якої визначають енергію активації реакції:
де m - відношення масштабу по осі абсцис до масштабу по осі ординат. Відрізок, що відсікається продовженням прямої на осі ординат, відповідає ln/k0. Знаючи константу швидкості при двох значеннях температури, можна розрахувати енергію активації:
Енергію активації можна визначити і за виразом:
Вплив температури і енергії активації на швидкість хімічних реакцій можна виразити рівнянням Арреніуса в експоненціальному вигляді:
де Якщо концентрації реагуючих речовин дорівнюють 1 моль/л, то рівняння Арреніуса дає змогу виразити залежність швидкості реакції від температури:
Оскільки в рівнянні температура входить у показник ступеня, то швидкість хімічних реакцій значною мірою залежить від зміни температури. Згідно з емпіричним правилом Вант-Гоффа: підвищення температури на кожні 10 градусів збільшує швидкість реакції приблизно в 2 - 4 рази. У математичній формі правило Вант-Гоффа записується так:
де Температурний коефіцієнт можна знайти за виразом:
Рівняння Вант-Гоффа є приблизним, оскільки швидкість реакції, крім температури залежить також від енергії активації, яка в свою чергу, залежить від температури. Молекулярність і порядок хімічних реакцій. Молекулярніть
|
|||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-29; просмотров: 113; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.14.249.191 (0.011 с.) |



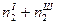 ;
;
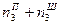 ;
;

 ;
;

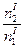
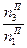
 ;
;



 ;
;
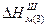
 .
.



 (2.16)
(2.16)
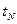 до
до  (лінія процесу NL) утворюється пересичення
(лінія процесу NL) утворюється пересичення

 у важкорозчинну сполуку СаСО3 (
у важкорозчинну сполуку СаСО3 ( = 4,84 . 10-9) з утворенням пересиченого розчину (200 - 300 мг/дм3 СаСО3).
= 4,84 . 10-9) з утворенням пересиченого розчину (200 - 300 мг/дм3 СаСО3).
 за певних умов випадатиме в осад, доки йонний добуток не буде дорівнювати
за певних умов випадатиме в осад, доки йонний добуток не буде дорівнювати 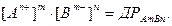
 навпаки при введенні в розчин сполука розчинятиметься до настання вищезгаданої рівноваги.
навпаки при введенні в розчин сполука розчинятиметься до настання вищезгаданої рівноваги. (2.17)
(2.17) (2.18)
(2.18)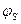 дорівнює:
дорівнює: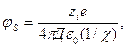 (2.19)
(2.19) - діелектрична проникність вакууму;
- діелектрична проникність вакууму;  - ефективний радіус йонної сфери (уподібнений радіусу провідної сфери із зарядом, що дорівнює заряду йонної сфери);
- ефективний радіус йонної сфери (уподібнений радіусу провідної сфери із зарядом, що дорівнює заряду йонної сфери);  - константа, яка дорівнює:
- константа, яка дорівнює: (2.20)
(2.20) :
: (2.21)
(2.21) - коефіцієнт активності.
- коефіцієнт активності. (2.22)
(2.22) (2.23)
(2.23)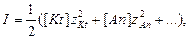 (2.24)
(2.24) - заряди відповідно катіонів і аніонів.
- заряди відповідно катіонів і аніонів. (2.25)
(2.25)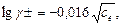 (2.26)
(2.26) ) дорівнює:
) дорівнює: (2.27)
(2.27) ) і дво-одновалентних (
) і дво-одновалентних ( :
: (2.28)
(2.28) ):
): (2.29)
(2.29) (2.30)
(2.30) дорівнює:
дорівнює:






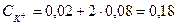 моль/л або 180 моль/м3.
моль/л або 180 моль/м3.




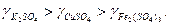


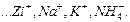


 (2.31)
(2.31) і
і  - відповідно розчинність і коефіцієнт активності при І = = 1.
- відповідно розчинність і коефіцієнт активності при І = = 1. утворюють прості і складні солі, розчинні комплексні сполуки, які знаходяться між собою в рівновазі.
утворюють прості і складні солі, розчинні комплексні сполуки, які знаходяться між собою в рівновазі.




 :
: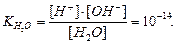










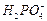 і
і 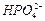 . Таким чином, загальний вміст Фосфору в грунтовому розчині (СР) визначається за рівнянням:
. Таким чином, загальний вміст Фосфору в грунтовому розчині (СР) визначається за рівнянням:



 - коефіцієнти активностей відповідно одно-, дво-, трьохзарядних йонів ортофосфатної кислоти.
- коефіцієнти активностей відповідно одно-, дво-, трьохзарядних йонів ортофосфатної кислоти. маємо:
маємо:


 - активність йонів Гідрогену (визначається із значень рН), К2 = 6,3 . 10-8 – друга константа дисоціації Н3РО4,
- активність йонів Гідрогену (визначається із значень рН), К2 = 6,3 . 10-8 – друга константа дисоціації Н3РО4,  і
і  - корелюючи фактори, які встановлюють частку вмісту йонів
- корелюючи фактори, які встановлюють частку вмісту йонів  і
і  в загальному вмісту Фосфору в розчині.
в загальному вмісту Фосфору в розчині.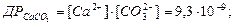






 , а також за допомогою матеріального балансу ортофосфатних компонентів розчину
, а також за допомогою матеріального балансу ортофосфатних компонентів розчину



 , рівняння матеріального балансу розчинності
, рівняння матеріального балансу розчинності  набуде вигляду:
набуде вигляду: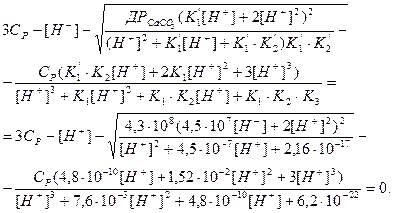
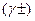 за рівнянням теорії Дебая-Гюккеля і порівняти їх з довідковими.
за рівнянням теорії Дебая-Гюккеля і порівняти їх з довідковими.
 (3.1)
(3.1)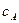 і
і 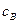 - концентрації реагуючих речовин; а і b - їх стехіометричні коефіцієнти. Правило, виражене рівнянням, називають основним постулатом хімічної кінетики.
- концентрації реагуючих речовин; а і b - їх стехіометричні коефіцієнти. Правило, виражене рівнянням, називають основним постулатом хімічної кінетики.
 реагентів А (1) і
D (2) з часом
реагентів А (1) і
D (2) з часом  для реакції aA+bB=cC+dD
для реакції aA+bB=cC+dD
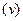
 можна ввести поняття про середню швидкість реакції
можна ввести поняття про середню швидкість реакції  (рис.3.2):
(рис.3.2): (3.2)
(3.2) і
і  - концентрація реагенту А в початковій
- концентрація реагенту А в початковій  і кінцевий
і кінцевий 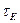 момент часу.
момент часу.
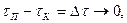
 (3.3)
(3.3) (3.4)
(3.4) (3.5)
(3.5)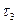 ) проводять дотичну. Тангенс кута нахилу
) проводять дотичну. Тангенс кута нахилу  дотичної до кривої відповідає швидкості реакції.
дотичної до кривої відповідає швидкості реакції.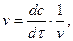 (3.6)
(3.6) - об’єм реакційного елемента.
- об’єм реакційного елемента. поділу фаз (каталізатора), яка в момент взаємодії реагентів практично не змінюється. Тому швидкість гетерогенних реакцій описується рівнянням:
поділу фаз (каталізатора), яка в момент взаємодії реагентів практично не змінюється. Тому швидкість гетерогенних реакцій описується рівнянням: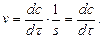
 не залежить від концентрації реагуючих речовин. Його фізичний зміст можна знайти, якщо прийняти, що концентрації реагуючих речовин А і В дорівнюють одиниці, тобто
не залежить від концентрації реагуючих речовин. Його фізичний зміст можна знайти, якщо прийняти, що концентрації реагуючих речовин А і В дорівнюють одиниці, тобто
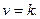
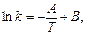 (3.7)
(3.7)
 (3.8)
(3.8)
 (3.9)
(3.9)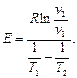 (3.10)
(3.10) (3.11)
(3.11) - передекспоненціальний множник, пропорційний числу зіткнень молекул.
- передекспоненціальний множник, пропорційний числу зіткнень молекул. (3.12)
(3.12) (3.13)
(3.13)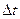 - збільшення температури;
- збільшення температури;  і
і  - швидкість реакції до
- швидкість реакції до  і після підвищення температури до
і після підвищення температури до  ;
; 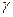 - температурний коефіцієнт швидкості реакції
- температурний коефіцієнт швидкості реакції 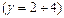 .
. (3.14)
(3.14)


