
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Тема 6. Фізико-хімія дисперсних систем та розчинів високомолекулярних сполукСодержание книги
Поиск на нашем сайте
Основи термодинаміки дисперсних систем. Дисперсними називають системи, які складаються з великої кількості диспергованих частинок – дисперсної фази, розподілених у рідкої, твердої або газовій фазах – дисперсійного середовища. Дисперсні системи можуть бути одержані або диспергуванням твердих речовин, або утворенням твердої фази із розчинів (конденсаційно кристалізаційні і коагуляційні структури). Розміри і форма часточок визначально впливають на властивості дисперсних систем. У більшості випадків утворення дисперсних систем потребує витрати роботи: або у вигляді механічної, або за рахунок процесів, що відбуваються в самій системі (в тому числі хімічних). Дисперсні системи, що виникли таким чином, є термодинамічно нерівноважними. Для свого скільки-небудь тривалого існування вони потребують спеціальної стабілізації. Такі термодинамічно нерівноважні дисперсні системи називають ліофобними. На відміну від ліофобних існує клас дисперсних систем, які виникають внаслідок самовільного диспергування макроскопічної фази, тобто без витрати механічної роботи, що підводиться зовні. Такі системи називають ліофільними. Вони є термодинамічно рівноважними і не потребують будь-якої додаткової стабілізації. Виникнення дисперсної системи внаслідок утворення частинок, що зародилися у новій стабільній фазі, можливе в любій метастабільній системі. Метастабільність, яка пов’язана з віддаленням від меж рівноважних умов існування даної системи, може бути викликана як відхиленням в хімічному складі фаз (пересичення), так і внаслідок фізико-хімічного впливу на систему (зміна тиску або температури).
Розглянемо процес утворення дисперсної системи шляхом диспергування макрофаги на прикладі відщеплення одної сферичної частинки радіусом r від макрофаги І, яка знаходиться в області стабільності цієї фази (рис.6.1). На рисунку цьому процесу відповідає перехід від стану І до стану ІІІ. Роботу утворення (диспергування) частинки (Wd) можна виразити як:
де SK – поверхня критичного зародку. Різниця між хімічними потенціалами речовини частинки
де N – кількість речовини (в молях), що утворює зародок; V – молярний об’єм речовини в зародку. Утворення сферичної частинки (ІІІ) можливе також із деякої макроскопічної фази ІІ, стан якої відрізняється від стану макрофаги. Їх хімічні потенціали різні, тому
Робота утворення цієї частинки, яка пов’язана зі зміною фазового стану
В той же час
де
Таким чином, робота утворення сферичної частинки при фазовому перетворенні має вираз:
З цього виразу випливає, що величина роботи У випадку рівності хімічних потенціалів:
вираз (6.4) приймає вигляд:
Цей вираз вперше отримав Гіббс. Як бачимо з виразу:
тоді як Колоїдно-дисперсні системи. Мікрогетерогенні (колоїдні) системи займають проміжковий стан між макроскопічними гетерогенними системами і молекулярними розчинами – гомогенними системами. Колоїдно-дисперсні системи характеризуються розміром частинок дисперсної фази. Чим дрібніші часточки і чим більше вони витягнуті, тим сприятливіші умови для виникнення міжчасткових контактів і колоїдного структуроутворення. Проте диспергування зумовлює надзвичайно велику вільну поверхневу енергію часточок. Залежність сумарної поверхневої енергії (
Класифікація дисперсних систем. В основі класифікації дисперсних систем лежать три загальні ознаки: дисперсність, гетерогенність та питома поверхня. За ступенем дисперсності дисперсні системи поділяють на три типи: грубодисперсні (макрогетерогенні і мікрогетерогенні), колоїдно-високодисперсні і молекулярно-дисперсні (табл.6.1). Цей поділ зумовлений розмірами частинок дисперсної фази. Класифікація дисперсних систем за агрегатним станом фаз (дисперсної фази і дисперсійного середовища) охоплює більшу частину різноманіття колоїдів і дає змогу виділити 9 (або строго кажучи, 8) типів дисперсних систем (табл.6.2). В неї агрегатні стани фаз позначають літерами: Т – тверда; Р – рідка; Г – газоподібна, а дисперсні системи – як дроби, де чисельник – дисперсна фаза, знаменник – дисперсійне середовище. У випадку, коли агрегатні стани фаз однакові до літер дробу добавляють індекси 1 – агрегатний стан дисперсної фази, а 2 – дисперсійного середовища (Т1/T2, P1/P2). З наведеної класифікації (табл.6.2) випливає, що дисперсні системи, в яких дисперсійне середовище газоподібне або рідке, за кінетичними властивостями частинок дисперсійної фази можна віднести до вільно дисперсних систем (золі), тобто у яких дисперсна фаза рухома. До них відносяться аерозолі, емульсії, ліозолі. Дисперсні системи з твердим дисперсійним середовищем, у якому частинки дисперсної фази можуть вільно пересуватися, а також висококонцентровані суспензії (бетонне тісто, глиняні пасти) і піни відносяться до зв’язанодисперсних систем (драглі). Зв’язанодисперсні системи містять у своїй структурі мікропори (2 - 200 нм) і макропори (порожнечі, канали) (> 200 нм), тобто являють собою високопористі тіла. Таблиця 6.1
Класифікація дисперсних систем залежно від розмірів частинок дисперсної фази
Таблиця 6.2
Класифікація дисперсних систем за агрегатним станом фаз
Колоїдно-дисперсні системи з рідким дисперсійним середовищем (ліозолі) називають колоїдними розчинами або просто золями. Якщо дисперсійним середовищем є вода такі золі називаються гідрозолями, а у випадку органічних рідин – органозолями, тобто якщо спирт – алкозолем, якщо етер – етерозолем і т.п. Необхідною умовою утворення дисперсних систем типу Р1/Р2, які називаються емульсіями, є їх утворення із взаємно нерозчинних або малорозчинних рідин. Дисперсні системи типу Г1/Г2 відсутні. Проте навіть в цьому випадку слід приймати до уваги специфічну мікронеоднорідність суміші газів, що обумовлена флуктуацією густини. Саме наявністю флуктуації густини і розсіюванням на неї світла пояснюється голубий колір неба: якби атмосфера була зовсім однорідною („оптично порожньою”), то небо було б чорним. В дисперсних системах частинки однієї дисперсної фази можуть мати міцний зв’язок з дисперсійним середовищем, а другої – слабкий. За цією ознакою колоїдно-дисперсні системи поділяються на два класи: ліофільні, колоїдні частинки які значно взаємодіють з дисперсійним середовищем, та ліофобні, колоїдні частинки які взаємодіють мало або практично зовсім не взаємодіють. Ліофільні колоїдно-дисперсні системи, дисперсійним середовищем яких є вода, називають гідрофільними, а ліофобні – гідрофобними. Типовими прикладами гідрофільних колоїдних систем є суспензії глини (золі гідроксидів металів), а гідрофобних – колоїдні розчини золота (Ауруму), сульфідів металів. Ліофільні системи утворюються шляхом довільного диспергування, отже, вони є термодинамічно стійкими. Ліофобні системи утворюються з перенасичених систем або шляхом диспергування великих частинок, внаслідок чого вони термодинамічно нестійкі. Тому гідрофільні золі добувають у значно більших концентраціях ніж гідрофобні; вони стійкіші до дії електролітів. Осади, які утворюють гідрофільні золі, дуже пухкі, об’ємні, що обумовлюються наявністю між частинками прошарків дисперсійного середовища внаслідок слабкої взаємодії між частинками. За топографічною ознакою частинок дисперсної фази дисперсні системи поділяються на корпускулярні, ламінарні (плівкові), фібрілярні (волокнисті). Наведена сукупність класифікацій надає можливість більш повніше характеризувати любу дисперсну систему, що розглядається, чого неможливо досягти при використанні любої окремої класифікації. Високомолекулярні сполуки та їх властивості. Високомолекулярні сполуки (ВМС), або полімери, містять у складі своїх молекул десятки і сотні тисяч (мільйонів) атомів. Побудовані вони з елементарних ланок, які повторюються, внаслідок взаємодії і з’єднання між собою однакових або різних молекул-мономерів. ВМС являють собою групу органічних речовин з специфічними властивостями, які притаманні тільки їм. Так, ВМС здатні до утворення термодинамічно рівноважних молекулярних розчинів з особливими термодинамічними властивостями, що обумовлені гнучкістю ланцюгів макромолекул, які володіють великою кількістю конформацій. В розчинах, в залежності від характеру взаємодії макромолекул, ВМС можуть існувати або у вигляді гнучких ланцюгів (статистичних клубків), або як щільні глобули згорнутих ланцюгів, або у вигляді асоціатів одної молекули з іншою. Встановлено, що розчини високополімерів гомогенні, тобто мають властивості справжніх розчинів низькомолекулярних сполук. Проте за величиною молекул розчини полімерів наближаються до колоїдних. Тому розчини ВМС мають деякі ознаки колоїдних розчинів. Синтетичні і природні ВМС. ВМС утворюються полімеризацією або поліконденсацією мономерів. Полімеризація відбувається з мономерами, які містять атоми з ненасиченими кратними зв’язками (СН2 = СН2, СН2 = С – СН = СН2). │ СН3 При цьому подвійний зв’язок між атомами кожної з молекул мономеру розривається і за рахунок таких зв'язків відбувається сполучення величезної кількості (n) молекул мономеру у великі молекули полімеру: n . СН2 = С – СН = СН2 │ │ СН3 СН3 (ізопрен) (поліізопрен (каучук))
Поліконденсація супроводжується виділенням низькомолекулярних речовин:
(терефталева кислота) (етиленгліколь)
(поліетилентерефталат)
Можливо також утворення нових високомолекулярних сполук з мономерів, внаслідок їх хімічного перетворення:
Можливі оцінки розчинності полімерів, взаємодії полімеру з розчинником і визначення межи розчинності до сих пір ще достатньо не вивчені. Встановлено, що хімічна і структурна спорідненість між полімером і розчинником полегшує розчинність. Високомолекулярні сполуки можуть утворювати і колоїдні розчини (дисперсії). В таблиці 6.3 приведені деякі представники полімерів і речовини, що їх розчинюють. В залежності від природи полімеру і розчинника і термодинамічних умов розчини полімерів в низькомолекулярних рідинах розглядають або як колоїдні системи, або як істинні розчини. ВМС, що утворені з двох-трьох і більше ланок різного складу, поєднують у собі властивості вихідних мономерів. Їх називають сополімерами. Прикладами сополімерів є сополімери стиролу з малеїновою кислотою
сополімери стиролу з бутадієном і акрилонітрилом
Таблиця 6.3
Високомолекулярні сполуки і речовини, що їх розчинюють
сополімери аценафтилену з вінілкарбазолом, стиролом, метакрилатом, вінілацетатом, ізобутиленом та ін. Їх застосовують в якості термостійких йоннообмінних смол, плівок, волокон, штучного грунту тощо. За допомогою ультразвукових коливань, електромагнітних вібраторів та іншими способами, ряд механічно подрібнених полімерів утворюють з ланцюгів своїх молекул радикали. Взаємодія між такими радикалами надає можливість одержання нових високополімерів. Високомолекулярні сполуки можуть мати як органічну, так і неорганічну природу. Для утворення ланцюгів полімерних сполук можуть служити не тільки Карбон або Силіцій, але і Алюміній, Бор, Магній, Фосфор, Тітан та ін. Наприклад, це поліфосфорні кислоти:
фосфоноксилоксани:
поліфосфіноборани:
гідраргіліт:
Значну кількість природних високомолекулярних сполук біологічного походження (вуглеводи, білки) також відносять до полімерів (біополімерів), наприклад целюлозу, крохмаль, полісахариди:
Клітини усіх тканин живих організмів і рослин та міжклітинна речовина також є біополімерами. Якщо полімери в основному або бічних ланцюгах містять йоногенні групи, їх називають поліелектролітами. До них відносяться желатина, казеїн, альбумін і альгінати. З синтетичних поліелектролітів до них відносяться поліакрилова кислота, поліакриламід і інші полімери, які містять кислотні (─CОО-, HOSO3-, ОРО3Н2-) або основні (NH3+, NHC(NH2)2+, N(CH3)3+) групи. До полікислот відносяться нуклеїнові кислоти, більшість муколісахаридів, гіалуронова, альгінова і карагінова кислоти, гуміарабік. Найпростіша синтетична поліакрилова полікислота у водному розчині слабко дисоціює:
Типовим представником поліоснов є полівініламін, йоннізація якого в кислому середовищі відбувається із захопленням протону:
Поліоснови, як і полікислоти сильніше йонізовані у сольовій формі:
Сполуки в яких в одному ланцюгу сполучаються кислотні і основні групи називаються поліамфолітами, наприклад:
До поліамфолітів відносяться усі білки. Поліамфоліти грають велику роль в природі: в склад біологічних мембран входять ліпіди, які містять амінні і кислотні залишки, що визначають їх амфотерні властивості. Основні особливості поліелектролітів зводяться до наступного. У воді їх макромолекули обмінюють протони і інші йонногенні групи на йони оточуючого середовища і таким чином набувають заряд. Так карбоксили полікислот в кислотній області рН залишаються практично нейоннізованими, а в нейтральній або лужній області з’являються заряджені групи – СОО-. Поліоснови, навпаки, залишаються нейонізованими в лужному середовищі і набувають позитивний заряд (NH3+) в кислому. Залежність заряду поліелектроліту від рН середовища представлена на рисунку 6.3.
У водних розчинах поліамфоліти дуже легко гідролізуються за рівнянням:
Основні групи – NH3ОН в молекулі
В кислому середовищі білкові молекули заряджаються позитивно:
Таким чином, знак зарядів амфотерних молекул білка залежить від реакції розчину (рис.6.3, б). Як видно з рисунка, точка перетину кривої з пунктирною лінією, відповідає нейтральному стану макромолекули в розчині. Такий ізоелектричний стан (рІ) у різних білків настає при різних концентраціях йонів Гідрогену розчину (табл.6.4). Таблиця 6.4 Значення рІ деяких білків
В ізоелектричній точці протилежно заряджені групи –NH3+ та –СОО- притягуються одна до одної, і молекула білка закручується у спіраль (рис.6.4, б).
При зміщенні рН від ізоелектричної точки в бік зменшення або збільшення рН однойменно заряджені групи відштовхуються, і молекула білка розпрямляється, набуваючи лінійної форми (рис.6.4, а, в). Будова ВМС. Будова молекул високополімерів залежить від будови молекул вихідних мономерів. Маромолекули, що утворюються при полімеризації, можуть мати як лінійну, розгалужену, сітчасту, так і більш складну форму (рис.6.5). Наприклад, молекули целюлози, крохмалю і глікогену складаються з глюкозних залишків (рис.6.5, в), які з’єднуються між собою через атоми Оксигену. Молекула целюлози має лінійну форму будови, а молекули крохмалю і глікогену складаються з розгалужених ланцюгів, а саме – молекула крохмалю має деревоподібну, а глікогену – кущоподібну форму (рис.6.6). Це пояснюється тим, що у процесі синтезу лінійне розташування ланок у ланцюгу може порушуватися, при цьому утворюються розгалуження. До утворення просторових або зшитих структур приводить введення природних або спеціальних домішків хімічно активних низькомолекулярних речовин. Наприклад, природний каучук, який характеризується суворо лінійним розташуванням ланок у ланцюгу, при вулканізації (вводиться Сульфур) набуває зшиту структуру. В макромолекулі полімеру ланки ланцюга сполучені між собою хімічними зв’язками, які навіть при незначному збільшенні відстані між сусідніми атомами Карбону різко послаблюються. Тому під впливом теплового руху взаємне розташування атомів Карбону може змінюватись. Завдяки цьому макромолекула може набувати різноманітних конформацій, тобто гнучкості.
Дуже гнучкі макромолекули прагнуть набути енергетично вигідну сферичну форму і утворювати глобули (рис.6.7, а). Глобули можуть утворюватися з одиничної макромолекули, або з кількох макромолекул (рис.6.7, б, в).
Макромолекули можуть складатися у пачки (рис.6.7, г, д, е, ж) утворювати великі і сильно витягнуті агрегати-фібрили (рис.6.7, з). Пластинчасті і голкоподібні структури полімерів можуть утворювати більш складніші і досконаліші структури-сфероліти і навіть монокристали. Властивості розчинів ВМС. Особливість розчинення ВМС полягає у тому, що розчинність полімерів можлива лише у тих розчинниках, до яких вони ліофільні, тобто гідрофільні полімери розчиняються у воді та інших полярних розчинниках, а гідрофобні полімери – у вуглеводнях. Так, желатина розчиняється у воді, а каучук – у бензені. Якщо полярність розчинника не співпадає з полярністю ВМС, в цьому випадку при розчиненні утворюються колоїдні розчини, а споріднені ВМС і розчинник утворюють істинні розчини. Наприклад, розчин каніфолі у воді є істинним розчином, а якщо до цього розчину додати спирту – утворюється колоїдний розчин (дисперсія каніфолі у спирті). Розчиненню полімеру передує набухання, тобто проникнення розчинника в порожнині полімеру. При цьому збільшуються маса і об’єм зразка полімеру ( Кінетика процесу набухання зразка полімеру приведена на рис.6.8. Кінетичні криві описують три стадії набухання: початкову (обмежену) стадію набухання (крива 1), подальшу стадію набухання з частковим розчиненням зразка полімеру (крива 2) і стадію необмеженого набухання з повним розчиненням зразка полімеру при якому макромолекули полімеру переходять у розчин. Ступінь набухання (
Набухання і розчинення ВМС – самовільний процес, що відбувається при зменшенні енергії Гіббса (ΔG<0). На процес впливають такі фактори: в’язкість розчинника, молекулярна маса полімеру, його надмолекулярна структура, температура розчинення. Велике значення має хімічна природа полімеру і розчинника. Так, набуханню і розчиненню сприяє утворення гідрогенних зв’язків між полярними групами полімеру і розчинника. Підвищення в’язкості розчинника зменшує швидкість проникнення рідини в полімер, а це значно сповільнює процес набухання. Навпаки підвищення температури прискорює процеси набухання і розчинення. Істотно впливає на процес розчинення зростання молекулярної маси полімеру. Більша довжина макромолекули потребує більшої витрати енергії на розрив усіх зв’язків в макромолекулі, тому розчинність зменшується. Надмолекулярна структура високополімерів також значно впливає на процеси набухання і розчинення ВМС. Так, у щільно згорнуті полімерні клубки альбуміну білків, глікогену молекули розчинника дуже важко проникають, тому такі глобулярні полімери практично не набухають. В той же час, якщо глобули полімерів мають велику спорідненість до розчинника, їх взаємодія послаблює зв’язки між окремими глобулами і вони переходять у розчин. Гірше і важче набухають і розчиняються полімери з розгалуженими мікромолекулами і кристалічні полімери. Перші мають між макромолекулами поперечні зшивки, які значно зменшують проникнення розчинника і повністю виключають можливість розчинення полімеру. Другі взаємодіють з розчинником лише поверхнею контакту, що обмежує набухання і розчинення кристалічного полімеру.
Набухання поліелектролітів у воді в значній мірі залежить від стану окремих ланок молекули білка. В ізоелектричній точці білка (рІ) протилежно заряджені групи –NH3+ та –СОО- притягуються і молекула білка закручується у спіраль (згортається). Проникнення розчинника у макромолекулу зменшується, тому в ізоелектричній точці набухання мінімальне. При зміні рН в кислий або лужний бік від ізоелектричної точки однойменно заряджені групи відштовхуються, і молекула білка розпрямляється, що призводить до зростання набухання. Залежність ступеню набухання білків від рН приведена на рис.6.9. Добування і властивості дисперсних систем і розчинів ВМС. Виникнення дисперсної системи в результаті утворення частинок нової стабільної фази в метастабільної системі може бути викликана як відхиленням в хімічному складі фаз, так і за рахунок фізико-хімічного впливу на систему. Це в першу чергу різні хімічні реакції, які приводять до виникнення високих концентрацій малорозчинних сполук, що обумовлює пересичення в системі, а також зміна тиску, температури і складу фаз. Процеси утворення дисперсних систем різної дисперсності і концентрації широко розповсюджені в природі і використовуються в різних областях технології. Так, окисно-відновні реакції лежать в основі багатьох методів добування золів, наприклад,
Важливе значення мають утворення гідрозолей в процесах гідролізу солей.
хімічна, або дисолюційна пептизація: Fe(OH)3 + HCl L FeOCl + 2H2O, обмінні реакції:
які можуть застосовуватися для добування дисперсних систем. Метод пептизації полягає у дезагрегації частинок осаду розпушеної структури, між якими є прошарок дисперсійного середовища. За механізмом пептизація поділяється на дисолюційну, адсорбційну і пептизацію промиванням розчинника (дисперсійним середовищем). Адсорбційна пептизація – це вибіркова адсорбція на осаді йонів, електролітів, що додаються до осадів. При хімічній, або дисолюційної пептизації речовини, що додаються до осадів, вступають в реакцію з поверхнею осадів. Промивання осадів розчинником або дисперсійним середовищем, створює вимивання надлишку електроліту і утворюється стійкий золь. Пептизація може відбуватися при додаванні до осаду неелектролітів, молекули яких адсорбуються на частинках осаду і запобігають їх злипанню. Такими речовинами є органічні кислоти, багатоатомні спирти, тощо. Для одержання колоїдних розчинів велике практичне значення мають методи диспергування, що грунтуються на подрібненні великих частинок до колоїдного стану. Процеси диспергування потребують великих енергетичних витрат. Для добування високодисперсних систем використовують колоїдні млини. Важливу роль в інтенсифікації процесів диспергування грає введення поверхнево-активних середовищ, що значно зменшує енергетичні витрати на ці процеси. Високодисперсні золі металів і сплавів у самих різних дисперсійних середовищах можуть бути одержані методом електророзпорушення, механічним або акустичним диспергуванням. Електричні явища. Завдяки надлишку поверхневої енергії, якою володіють дисперговані часточки, на межі розділу фаз на міжфазових поверхнях виникає взаємодія, що приводить до утворення подвійного електричного шару (ПЕШ). Просторове розділення зарядів і утворення ПЕШ характерно для любої межі двох фаз, в системі якої є йони і інші заряджені частинки. Причиною утворення ПЕШ і відповідного стрибка потенціалу можуть бути: а) обмін зарядженими частинками; б) вибіркова адсорбція йонів; в) адсорбція полярних молекул. Знак заряду поверхні частинок залежить від природи дисперсної фази та дисперсійного середовища. При змочуванні водою поверхня таких речовин, як метали, сульфіди металів, силікагель, крохмаль, деревина, папір, гуміарабік тощо заряджається негативно, а нерозчинних оксидів, гідроксидів металів, деяких солей – позитивно. На знак заряду поверхні частинок значно впливає значення діелектричної проникності. З двох спряжених фаз позитивно заряджається фаза з більшою діелектричною проникністю, наприклад, частинки глини у воді заряджені негативно, а вода – позитивно. Подвійний електричний шар складається з двох частин: щільної (внутрішня обкладка) і дифузної (зовнішня обкладка((рис.6.10). Більш близька до поверхні внутрішня обкладка ПЕШ (шар Штерна-Гельмгольца) відповідає частині, де адсорбційні сили суттєві. У більш віддаленій дифузній частині (шар Гуї-Чепмена) ними можна знехтувати.
Внутрішня обкладинка представляє собою адсорбційний монойонний шар, товщиною не менше двох радіусів йонів (б) (рис.6.11). За Штерном в неї знаходяться потенціаловизначаючі йони, фіксовані твердою поверхнею і частина протийонів адсорбційного шару. Зовнішня обкладинка складається з протийонів дифузного шару. Її товщина (λ) може бути значною і залежить від властивостей і складу дисперсної системи. Розрахувати її можна за рівнянням:
де На межі твердої та рідкої фаз виникає так званий поверхневий, або На межі адсорбційного і дифузного шарів (лінія АА) виникає стрибок потенціалу, який Штерн назвав
Заряд адсорбційного шару складається із зарядів йонів, що адсорбуються як за рахунок електростатичного а
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-29; просмотров: 188; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.119.253.198 (0.019 с.) |


 (6.1)
(6.1) і вихідної стабільної фази
і вихідної стабільної фази  в цьому випадку складає:
в цьому випадку складає: , (6.2)
, (6.2) .
. , дорівнює:
, дорівнює: .
. ,
, - “хімічна” робота
- “хімічна” робота  переносу N речовини із стану ІІ в стабільну фазу І:
переносу N речовини із стану ІІ в стабільну фазу І: (6.3)
(6.3) (6.4)
(6.4) на величину “хімічної” роботи
на величину “хімічної” роботи  (6.5)
(6.5) (6.6)
(6.6)
 компенсуються “хімічною” роботою
компенсуються “хімічною” роботою  , а рівність (6.5) є нестійкою рівновагою в умовах якої відбувається виникнення критичних зародків нової фази.
, а рівність (6.5) є нестійкою рівновагою в умовах якої відбувається виникнення критичних зародків нової фази.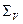 ) від ступеня дисперсності (
) від ступеня дисперсності ( ) часточок має екстремальний характер (рис.6.2). Ступінь дисперсності, що відповідає інтервалу
) часточок має екстремальний характер (рис.6.2). Ступінь дисперсності, що відповідає інтервалу  від
від 
 (- СН2 - С = СН – СН2 -)n.
(- СН2 - С = СН – СН2 -)n.


 С6Н5
С6Н5




 .
.





 а)
а)
 б)
б)

 дисоціюють слабкіше ніж кислотні – СООН, тому молекула білка має негативний заряд:
дисоціюють слабкіше ніж кислотні – СООН, тому молекула білка має негативний заряд:

 -Глобулін
-Глобулін




 ).
). ) при цьому набуває від’ємного значення (крива 3).
) при цьому набуває від’ємного значення (крива 3).







 , (6.6)
, (6.6) - діелектрична проникність дисперсійного середовища;
- діелектрична проникність дисперсійного середовища;  - діелектрична стала; І – йонна сила розчину; F – число Фарадея.
- діелектрична стала; І – йонна сила розчину; F – число Фарадея.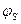 -потенціал. Величина
-потенціал. Величина  - потенціалом, або адсорбційним потенціалом. Потенціал у адсорбційному шарі зменшується лінійно (пряма MN) від
- потенціалом, або адсорбційним потенціалом. Потенціал у адсорбційному шарі зменшується лінійно (пряма MN) від 
 - потенціаловизначаючі йони;
- потенціаловизначаючі йони;  - протийони; заштрихована частина – агрегат
- протийони; заштрихована частина – агрегат



