Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Тема 5. Фізико-хімія поверхневих явищ. АдсорбціяСодержание книги
Поиск на нашем сайте
Поверхневі явища і їх значення. В любих гетерогенних системах, особливо у високодисперсних, природа межи між частинками дисперсної фази і дисперсійним середовищем, молекулярна будова межі поділу і фізико-хімічні взаємодії, що відбуваються на ній, визначають багато чисельні явища і процеси, які характерні для заданої системи. Вивчення поверхневих явищ відкриває шляхи регулювання цих явищ і взаємодію у природі, а також регулювання ними у техніці. Керування фізико-хімічними явищами на межі поділу дисперсних фаз дозволяє вирішити такі задачі, як закріплення грунтів (зниження їх осідання і утворення плавунів), тонко регулювати процеси структуроутворення в глинистих суспензіях, які застосовують при свердлуванні, забезпечувати рухливість і незлипання ниток, коли їх прядуть, знижувати водопотребу цементних розчинів і маслопотребу фарб, або, наприклад, забезпечувати добру адгезію бітуму до гравію, щебеню в асфальтобетоні. На межі поділу фаз у живому організмі і рослинах відбуваються біохімічні процеси обміну і синтезу речовин, ферментативні реакції і інші процеси, які пов’язані з життєдіяльністю організмів і рослин. Дисперсність. Питома поверхня і вільна поверхнева енергія. Дисперсність – характеристика розмірів частинок в дисперсних системах. За міру дисперсності речовини прийнято ступінь дисперсності
Дисперсність полідисперсних систем характеризується функцією розподілення маси (об’єму) частинок за їх розміром f(d) (рис.5.1). Крім того, вони характеризуються ще і ступенем полідисперсності, тобто розмірністю кривої розподілу – її основою d’min – d’max – повним інтервалом частинок і їх ймовірним діаметром d0. Розмір частинок визначає також іншу, дуже важливу характеристику дисперсної системи – питому поверхню SПИТ:
де S і V – відповідно сумарні поверхня і об’єм дисперсної фази; S’ і V’ – поверхня і об’єм одної часточки.
Кількість співвідношення між
SПИТ в табл.5.1 відповідає умові диспергування при збереженні форми і дворазового зменшення розмірів часточок. Величина питомої поверхні систем, наприклад, із куль одного розміру діаметром 1 мм SПИТ = 6 . 103 м-1, для часточок розміром 1 нм SПИТ = 6 . 109 м-1. Загальна поверхня 1 м3 дисперсної фази таких часточок (1 нм) становить 6000 км2. Велика міжфазна поверхня, яка притаманна дисперсним системам, визначає особливо важливу роль, яку грають в них явища, що відбуваються на межі поділу фаз, - поверхневі явища.
Таблиця 5.1 Співвідношення між
Дві фази, наприклад рідина і пара в однокомпонентній системі, можуть існувати в рівновазі тільки при наявності стійкої межі поділу між ними. Міжмолекулярні взаємодії молекул в площині фаз і в об’ємі рідкої фази (рис.5.2) істотно відрізняються за енергіями.
Молекула А, що міститься у поверхневому шарі на межі поділу фаз, перебуває під впливом молекулярних сил взаємодії з боку прилеглих до неї молекул, які розміщені як на поверхні поділу фаз, так і знизу під нею. Положення таких молекул енергетично нескомпенсоване (R >> 0). Якщо молекула знаходиться в об’ємі рідкої фази (В), вона рівномірно притягається з усіх боків такими же за природою молекулами. Силове поле міжмолекулярної взаємодії в цьому випадку повністю зрівноважується (R=0). Таким чином, поверхневі молекули мають надлишкову енергію. Ця енергія може перетворюватись на роботу, яка витрачається на переміщення молекул з поверхні поділу фаз у глибину рідини. Такий надлишок енергії називають вільною поверхневою енергією
По каркасній рамці з дроту (шириною L) вільно переміщується легка планка. Перевернутий каркас занурюють в мильний розчин з гліцерином. Прилад виймають і повертають в попереднє положення. При цьому між каркасом і планкою утворюється двобічна плівка з мильного розчину (заштрихована площа). На петлю в планці підвішують вантаж під дією якого планка опускається і розтягує мильну плівку. Робота, яка при цьому витрачається дорівнює:
де Р – вага вантажу; dh – відстань, на яку перемістилась планка. Площа, на яку збільшилась поверхня плівки, становить:
де множник „2” відповідає двобічності плівки. Збільшення вільної поверхневої енергії системи при цьому відбувається на:
Підставляючи в це рівняння значення dS маємо:
звідки
Таким чином, величина Дію поверхневого натягу можна наочно представити у вигляді сукупності сил, що стягують краї поверхні до центру. Ці сили зображені на рис.5.4 стрілочками-векторами; довжина стрілочок відображає величину поверхневого натягу, а відстань між ними відповідає прийнятій одиниці довжини.
Оскільки питома вільна поверхнева енергія і поверхневий натяг є синонімами відповідно в якості розмірності величини Підвищення температури супроводжується пропорційним послабленням сил міжмолекулярної взаємодії, тому при досягненні критичної температури (Т = ТКР) Кожна чиста рідина має сталий коефіцієнт поверхневого натягу (при Т = const), величина якого залежить від природи речовини. Він тим більше, чим більша полярність молекул рідини, він також залежить від здатності молекул утворювати водневі зв’язки та ін. Нижче наведені значення поверхневого натягу для деяких рідин на межі з повітрям при 293 К (табл.5.2). Таблиця 5.2 Поверхневий натяг для деяких рідин на межі з повітрям при 293 К
З наведених значень поверхневого натягу випливає, що величина поверхневого натягу води в порівнянні з поверхневим натягом інших рідин має найбільшу величину. У випадку незмішуваних рідин міжфазний натяг розраховують за правилом Антонова:
де З термодинаміки випливає, що будь яка гетерогенна система прагне зменшити поверхневу енергію:
за рахунок зменшення або Зменшення Отже, наявність у міжфазних шарів надлишкової вільної енергії приводить до різноманітних явищ (змочування, адгезії, адсорбції тощо). Когезія та адгезія. Когезія (зчеплення) – притягання між молекулами (атомами і йонами) в об’ємі даного тіла. Речовини в конденсованому стані (тверді і рідкі тіла) характеризуються високою когезією, а газоподібні – дуже малою когезією. Когезія характеризує ідеальну міцність тіла, що визначається силою зчеплення утворюючих його молекул, атомів, йонів. Для отримання зв’язку поверхневої енергії з енергією зчеплення молекул в об’ємі конденсованої фази введена величина WК, яка називається роботою, або енергією когезії. Ця величина може бути визначена як робота, яку необхідно здійснити в оборотному ізотермічному процесі розділення на дві частинки стовпчика одиничного перерізу. Оскільки в такому процесі утворюються дві поверхні одиничної площі, робота когезії дорівнює подвійному значенню поверхневого натягу:
За мікроскопічною теорією Гамакера і Де-Бура роботу когезії визначають як результат підсумовування дисперсійних взаємодій між молекулами, які містяться в двох полунескінченних об’ємах конденсованої фази, що розділені плоским перерізом шириною h (рис.5.5).
Величина енергії взаємодії фаз Umol дорівнює енергії взаємодії молекул, що знаходяться в нескінченно довгому циліндрі одиничного перерізу S над площиною Q1, з молекулами у всьому об’ємі, розташованому над площиною Q2. В цьому випадку роботу когезії розглядають як ту границю, до якої намагається величина Umol при зменшенні товщини перерізу h до розміру молекули b. Таким чином при:
де А – стала Гамакера (Дж). WK має місце для любих рідких фаз: полярних і неполярних. Її називають ще когезійною міцністю, або міцністю на розрив. Так, когезійна міцність для води при 298 К складає:
Адгезія (прилипання) – молекулярний зв’язок між поверхнями двох різнорідних твердих або рідких тіл (фаз), що стикаються. Термодинамічною характеристикою адгезії є спад вільної енергії на 1см2 поверхні адгезійного шва в ізотермічному оборотному процесі. Представимо собі дві конденсовані речовини (2 і 3) з поверхнею 1 см2 на межі з газом (повітрям) (1). Це можуть бути дві рідини або рідина і тверде тіло:
При суміщенні цих поверхонь, тобто нанесенні одної речовини на другу, відбувається адгезія. Так як система залишається двофазною, то залишається і міжфазний натяг (
Енергія Гіббса для вихідного і кінцевого станів дорівнює:
де
або
Величина Wа характеризує спорідненість контактуючих фаз, тобто ступінь насичення нескомпенсованих поверхневих сил при контакті; навпаки, міжфазна енергія На відміну від когезії, коли робота витрачається на подолання сил зчеплення між однорідними молекулами, робота адгезії Wа пов’язана з енергією взаємодії різнорідних молекул (атомів). Змочування та розтікання. Якщо на поверхню твердого тіла нанести краплину рідини, то у такої системи присутні три різних поверхні поділу фаз: між рідиною і твердим тілом, рідиною і парою (повітрям) і твердим тілом і парою з поверхневим натягом
Лінія перетину усіх трьох поверхонь поділу називається лінією змочування (замкнута лінія змочування утворює периметр змочування). Кут У випадку, коли краплина рідини нанесена на іншу рідину схема взаємодії з поверхнями буде мати вигляд (рис.5.7).
Розглядаючи поверхневі натяги як сили, що прикладені перпендикулярно до одиниці довжини периметру змочування і що діють по дотичній до відповідних поверхонь (рис.5.6, а) можна записати умову рівноваги цих сил:
або В залежності від значень крайового кута змочування розрізняють такі випадки: 1) 2) 3) крайовий кут змочування не встановлюється – повне змочування поверхні, або розтікання. У відповідності з рівнянням Юнга (5.9) Рушійна сила процесу розтікання називається роботою розтікання WР (або коефіцієнтом розтікання). Величина WР дорівнює:
Робота розтікання може бути визначена як різниця робот адгезії і когезії:
Таким чином, добре змочування і розтікання можливі при великій роботі адгезії і при низькій роботі когезії. Якщо трьохфазна система складається із трьох рідких фаз, що не змішуються, можливі наступні випадки (рис.5.8):
Повне змочування одної рідини в другій (а), часткове змочування цих рідин (б) і коли рідини не змочуються (в). Якщо Таблиця 5.3 Коефіцієнти розтікання для деяких систем
У відповідності з (5.10) вуглеводи і інші органічні речовини з малим поверхневим натягом добре змочують практично усі тверді тіла. Вода-рідина з високою роботою когезії – добре змочує оксиди і розтікається на деяких силікатах, але не змочує флюорорганічні полімери, парафін. В першому випадку ці поверхні гідрофільні, а в другому – гідрофобні по відношенню до полярних молекул води. При вибірковому змочуванні, тобто коли кожна з рідин окремо змочує поверхню, встановлюється такий крайовий кут, який відповідає доброму змочуванню тією рідиною, спорідненість якої з цією твердою поверхнею більше. Якщо вода добре, ніж карбон гідроген, вибірково змочує поверхню, тобто До речовин з гідрофільною поверхнею відносяться: кварц, скло, оксиди, гідроксиди металів окисненні мінерали та ін. До гідрофобних об’єктів відносяться карбонгідрогени, флюориди, листя рослин, хітиновий покрив комах, шкіра тварин тощо. Важливою кількісною характеристикою енергетики змочування є теплота змочування – кількість енергії, що виділяється при змочуванні одиниці поверхні твердого тіла. Тому, що ентальпія поверхневого шару на межі тверде тіло-газ більше, ніж на межі тверде тіло-рідина, то при змочуванні завжди виділяється тепло, яке дорівнює різниці повних поверхневих енергій або ентальпій шарів після змочування (Н2) і до нього (Н1):
Теплота змочування залежить від кількості рідини, що насичена на поверхні того чи іншого тіла. Це обумовлено проникненням поля поверхневих явищ всередину на певну глибину, яка дорівнює товщині поверхневого шару у фазі рідини. На змочування твердих тіл рідинами великий вплив здійснює стан поверхні твердого тіла, а саме його шорсткість (кривизна поверхні). Згідно Дерягіну, у випадку контакту рідини з поверхнею реального твердого тіла вираз роботи адгезії необхідно записати у вигляді:
де Капілярність. Для високодисперсних систем характерна велика кривизна поверхні поділу фаз (шорсткість), тому необхідно враховувати її вплив при розгляданні термодинамічних властивостей таких систем. Розглянемо поведінку рідини у тонкому капілярі, який занурений в рідину. В цьому випадку можна рахувати, що меніск має сферичну форму (рис.5.9). При умові змочування рідиною стінок капіляру (
Величина
За рахунок
де Радіус кривизни меніска r пов’язаний з радіусом капіляра співвідношенням:
Висота капілярного підняття визначається за формулою Жюрена:
З рівняння (5.14) випливає, що чим краще рідина змочує стінки капіляра, тим вище підняття в ньому при даному значенні Капілярні явища мають місце в грунтах. Сукупність капілярів (пор) різних діаметрів в грунті над поверхнею ґрунтових вод утворюють капілярну кайму (рис.5.10). Форма кайми залежить від неоднорідності пор за розмірами. В піщаних грунтах вона більш виразна; при великій нерівномірності пор за розмірами (суглинисті грунти) капілярна кайма розтягнута по висоті. Висота кайми є одною із воднофізичних констант грунту і називається максимальною висотою капілярного підняття hК. Для піщаних грунтів вона складає приблизно 0,4-1 м, супіщаних – 1-1,5 м, суглинистих – 1,5-2,5 м, глинистих – 2,5-5 м. Характерним проявом капілярного тиску є явище виникнення капілярної стягуючої сили (F) між частинками при наявності меніска (рис. 5.11, а).
Рис.5.10. Розділ ґрунтової вологі по висоті: 1 – тверда фаза; 2 – ґрунтова вода; 3 – гідроскопічна волога; 4 – плівкова волога; 5 – капілярно-підвищена волога (капілярна кайма); 5а – капілярно-підперта волога; 6 – підвищена волога; 7 – ґрунтове повітря Величина цієї сили суттєво залежить від кількості рідини в меніску. Наприклад, при висиханні (рис.5.11, а) стягуюча сила максимальна. При збільшенні рідини до утворення циліндричного меніску капілярна стягуюча сила зменшується (рис.5.11, б). Коли утворюється „меніск” з параметрами r=2r0 ця стягуюча сила зникає, тобто F=0 (рис.5.11, в). Власне цим обумовлені факти „розпливання” сильно зволоженого піску і його більш менш доброї формозміни при слабкому зволоженні.
Капілярні сили в більшості визначають зчеплення частинок і безпосередньо зв’язані з цим зчепленням механічні властивості грунтів, різних технічних, харчових, лікувальних і інших порошків і паст. Велике значення капілярності має місце в просочуванні рідинами пористих тіл неорганічного походження (піднімання вологи по каменю фундаменту, цеглі). Капілярність відіграє велику роль у зберіганні і переміщення вологі в грунті. Руйнуючи капіляри розпушуванням грунту, припиняють піднімання води на поверхню і уповільнюють висихання грунту. А коткування грунту збільшує прилив вологи на поверхню. Адсорбція. Прагнення вільної поверхневої енергії (енергії Гіббса) набути мінімально можливого значення може задовольнятися не тільки зменшенням поверхні поділу фаз (
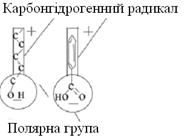
Проходження вільного процесу відбувається за рахунок утворення сферичної і гладкої рідкої поверхні, укрупнення частинок дисперсної фази (зменшення поверхні поділу фаз). До зменшення поверхневого натягу приводять теплові, електричні, фізико-хімічні явища (адсорбція та ін.), що відбуваються на поверхні поділу фаз. Адсорбція – концентрування речовини (адсорбата) із об’єму фаз на поверхні поділу між ними, наприклад, із газу або розчину на поверхні твердого тіла (адсорбента) або рідини. Адсорбційні сили при фізичній адсорбції мають ту ж саму природу, що і молекулярна взаємодія в газах, рідинах і твердих тілах. Адсорбція на межі поділу рідина-газ. Адсорбція залежить від природи розчинника і розчиненої речовини, особливо від значень величин поверхневого натягу цих речовин. Якщо поверхневий натяг розчинника, наприклад води, більший від поверхневого натягу розчиненої речовини (органічна речовина),то остання проявляє поверхневу активність, тобто зменшує поверхневий натяг розчину (рис.5.12). Речовини, які зменшують поверхневий натяг межі поділу фаз, називаються поверхнево-активними (ПАР). ПАР поділяють на аніонні, катіонні, нейонні, амфолітні. Прикладом таких ПАР є карбонгідрогени, ароматичні сполуки, солі жирних кислот, аміни та їх похідні, естери, білки тощо. ПАР – органічні сполуки дифільної природи. Їх будову та орієнтацію на воді схематично зображують кільцем з зігзагом або рискою (рис.5.13).
Рис.5.12. Зменшення поверхневого натягу від концентрації розчину на межі поділу вода-повітря і вода-карбонгідроген Оскільки полярні групи дифільних молекул ПАР мають велику спорідненість до води, тому на межі полярного і неполярного середовища молекули ПАР орієнтуються полярними групами до води, а неполярні (карбонгідрогенний радикал) виштовхуються у газову фазу (повітря).
Молекули ПАР мають досить великі, але слабко взаємодіючі між собою неполярні частини, тому завжди:
Внаслідок цього молекули ПАР частково (рис.5.13, а) або повністю (5.13, б) витискують із поверхневого шару полярні молекули води, тобто відбувається адсорбція ПАР. Величина адсорбції дорівнює різниці концентрацій адсорбату в поверхневому шарі і в об’ємі межуючих фаз. Вона позначається літерою Г і вимірюється кмоль/м2. Механізм процесу адсорбції описує рівняння Гіббса:
де С – загальна концентрація розчину, моль/м3; R – газова стала 8,314 Дж/моль К; Т – абсолютна температура, К;
Визначається G в Дж.м/моль або Н.м2/моль. Якщо поверхневий натяг розчину зменшується при збільшенні концентрації розчиненої речовини, тобто Позитивна та негативна адсорбція має місце в біологічних об’єктах живих організмів і рослин. Завдяки присутності в клітинних і міжклітинних рідинах поверхнево-активних речовин їх поверхневий натяг значно менший, ніж води, а це забезпечує їх проникність крізь біологічні мембрани, підсилює обмін речовин. Для визначення адсорбції ПАР на межі рідина-газ користуються ізотермою поверхневого натягу (рис.5.14, крива 1). До довільно вибраній точці на кривій поверхневого натягу проводять дотичну до перетину її з віссю ординат. Із побудування випливає:
звідси
Рис. 5.14. Залежність поверхневого натягу (1) та адсорбції (2) від концентрації ПАР на межі рідина-газ
За результатами декількох обчислень будують ізотерму адсорбції (рис.15.14, крива 2). Аналіз ізотерми абсорбції свідчить, що при низьких концентраціях адсорбція пропорційна концентрації, а при високих концентраціях досягає свого граничного значення (Гmax) і потім не змінюється. Досліджуючи залежність поверхневого натягу від концентрації водних розчинів органічних речовин біолог Дюкло і фізіолог Траубе встановили правило: поверхнева активність дифільних сполук у водних розчинах однакової концентрації збільшується у 3-3,5 разів зі збільшенням довжини карбонгідрогенного радикалу на одну – СН2 -групу, тобто
де n – число атомів Карбону в радикалі. Як видно з рис. 5.15, на якому зображені криві
Рис.5.15. Залежність поверхневого натягу від концентрації водних розчинів кислот Із правила Дюкло-Траубе випливає, що площа, яка припадає на одну молекулу максимально насиченого ПАР адсорбційного шару, є сталою в межах цього гомологічного ряду, а це можливо тільки при горизонтальному розташуванні карбогенгідрогенних радикалів на поверхні (див.рис.5.13, б). Для обчислення поверхневого натягу жирних кислот (або інших ПАР) з невеликим числом атомів Карбону (до С8) застосовують рівняння Шишковського (1908 р.):
де В – константа (однакова для гомологічного ряду ПАР); 1/A – константа, яка залежить від довжини карбогенгідроненного радикалу. За допомогою рівняння Шишковського (5.18) можна перейти від рівняння Гіббса (5.16) до рівняння ізотерми адсорбції Ленгмюра, яке має вигляд:
де Гmax – константа, що характеризує граничну концентрацію речовини в поверхневому шарі (див.рис.5.14); b – константа. Константу рівняння Ленгмюра можна визначити графічним методом (рис.5.16), перетворивши рівняння Гіббса на рівняння прямої:
На графіку пряма відсікає на осі ординат відрізок, який дорівнює Досліди Ленгмюра з жирними кислотами (від 14 до 34 атомів Карбону) показали, що незалежно від довжини молекули, площа, яка припадає на 1 молекулу в насиченому моношарі постійна, а це вказує на те, що молекули дійсно орієнтуються вертикально до поверхні (частокол Ленгмюра).
Разом з тим встановлено, що деякі молекули орієнтуються в адсорбційних шарах горизонтально, а площа, зайнята молекулою, зростає із збільшенням її довжини. Це обумовлено наявністю в молекулах декількох полярних груп. В таблиці 5.4 наведені величини площі (S) для деяких гомологічних рядів і окремих сполук. Адсорбція на межі поділу тверде тіло-газ. Система тверде тіло-газ належить до нерухомих меж поділу фаз. Тверді тіла – адсорбенти при контакті з газовою фазою можуть поглинати своєю поверхнею молекули газів і парів. Такий процес називається сорбцією. У випадку коли процес сорбції відбувається тільки поверхнею твердого тіла, тобто сорбовані молекули не розподіляються по його об’єму, то таке явище називають адсорбцією. Речовину, яка адсорбується називають адсорбтивом. Таблиця 5.4 Площі молекул для деяких гомологічних рядів і окремих сполук
Взаємодія між частинками адсорбтиву і адсорбента може мати різний характер. Залежно від природи адсорбційних сил розрізняють два види адсорбції: фізичну та хімічну. Хімічна адсорбція (хемосорбція) з
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-29; просмотров: 437; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.149.235.190 (0.012 с.) |

 . Це величина, обернена розміру (діаметру) часточки d:
. Це величина, обернена розміру (діаметру) часточки d: , (м-1). (5.1)
, (м-1). (5.1) (м-1) (5.2)
(м-1) (5.2)
 і SПИТ для часточок різних форм залежать тільки від розміру часточок. Наприклад, часточка сферичної форми (куляста) характеризується розміром діаметру d (табл. 5.1). Для неї:
і SПИТ для часточок різних форм залежать тільки від розміру часточок. Наприклад, часточка сферичної форми (куляста) характеризується розміром діаметру d (табл. 5.1). Для неї:







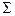 . Відповідна величина, віднесена до одиниці поверхні, називається питомою вільною поверхневою енергією
. Відповідна величина, віднесена до одиниці поверхні, називається питомою вільною поверхневою енергією 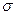 . Її можна розглядати як силу, яка протидіє збільшенню площі поверхні. Існування цієї сили наочно ілюструє дослід Дюпре (рис.5.3).
. Її можна розглядати як силу, яка протидіє збільшенню площі поверхні. Існування цієї сили наочно ілюструє дослід Дюпре (рис.5.3).

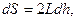
 (5.3)
(5.3)


 , Дж/м2
, Дж/м2
 (5.4)
(5.4)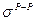 - міжфазний натяг на межі поділу першої рідини у другій, та другої у першій;
- міжфазний натяг на межі поділу першої рідини у другій, та другої у першій;  - поверхневий натяг на межі з газовою фазою рідини 2 у рідині 1 та рідини 1 у рідині 2 відповідно.
- поверхневий натяг на межі з газовою фазою рідини 2 у рідині 1 та рідини 1 у рідині 2 відповідно.

 (5.5)
(5.5)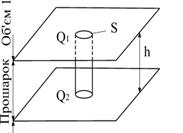

 (5.6)
(5.6) Дж/м2.
Дж/м2.
 ). В цьому процесі початкова енергія Гіббса зменшиться на величину, яка дорівнює роботі адгезії, тобто:
). В цьому процесі початкова енергія Гіббса зменшиться на величину, яка дорівнює роботі адгезії, тобто: . (5.7)
. (5.7)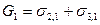 і
і 
 відповідно поверхневі натяги другого і третього конденсованих тіл з газом (повітрям) і міжфазний натяг на межі другого тіла з третім. Тоді
відповідно поверхневі натяги другого і третього конденсованих тіл з газом (повітрям) і міжфазний натяг на межі другого тіла з третім. Тоді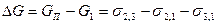
 (5.8)
(5.8) відповідно. Краплина рідини на поверхні твердого тіла представляє собою шаровий сегмент з висотою Н, радіусом кривизни r і радіусом кола трьохфазного контакту (периметр змочування) r1 (рис.5.6, а).
відповідно. Краплина рідини на поверхні твердого тіла представляє собою шаровий сегмент з висотою Н, радіусом кривизни r і радіусом кола трьохфазного контакту (периметр змочування) r1 (рис.5.6, а).
 між поверхнями рідина-пара і тверде тіло-рідина називається крайовим кутом змочування.
між поверхнями рідина-пара і тверде тіло-рідина називається крайовим кутом змочування.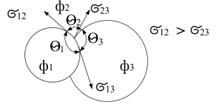

 (5.9)
(5.9) - поверхня змочується (або частково змочується);
- поверхня змочується (або частково змочується);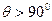 - поверхня не змочується (або погано змочується);
- поверхня не змочується (або погано змочується); або змочуванню відповідає умова
або змочуванню відповідає умова  (рис.5.6, а), незмочуванню -
(рис.5.6, а), незмочуванню -  (рис.5.6, б), а розтіканню -
(рис.5.6, б), а розтіканню -  .
.
 (5.10)
(5.10)
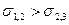 : а – повне змочування; б – часткове змочування; в – незмочування
: а – повне змочування; б – часткове змочування; в – незмочування
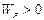 , то рідина буде розтікатися по поверхні, тому що цей процес супроводжується зменшенням вільної поверхневої енергії і відбувається самодовільно. У табл.5.3 наведені приклади, які пояснюють сказане.
, то рідина буде розтікатися по поверхні, тому що цей процес супроводжується зменшенням вільної поверхневої енергії і відбувається самодовільно. У табл.5.3 наведені приклади, які пояснюють сказане. , Дж/м2
, Дж/м2
 ,
Дж/м2
,
Дж/м2


 (5.11)
(5.11) - коефіцієнт шорсткості;
- коефіцієнт шорсткості;  і
і  - реальна і ідеальна поверхня твердого тіла.
- реальна і ідеальна поверхня твердого тіла. ) її поверхня буде викривленою з від’ємним радіусом кривизни r (вгнутий меніск). Внаслідок вгнутої поверхні меніска тиск на нього з боку атмосфери зменшується на силу
) її поверхня буде викривленою з від’ємним радіусом кривизни r (вгнутий меніск). Внаслідок вгнутої поверхні меніска тиск на нього з боку атмосфери зменшується на силу 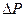 , яка дорівнює:
, яка дорівнює: (5.12)
(5.12) - різниця тисків у сусідніх фазах, що розділені викривленою поверхнею, називається капілярним тиском.
- різниця тисків у сусідніх фазах, що розділені викривленою поверхнею, називається капілярним тиском.
 (5.13)
(5.13) і
і  - густина рідини і її насиченої пари (або повітря); Н – висота підйому рідини; q – прискорення сили тяжіння.
- густина рідини і її насиченої пари (або повітря); Н – висота підйому рідини; q – прискорення сили тяжіння.
 (5.14)
(5.14) . За цією формулою можна підрахувати, що в капілярі радіусом 0,01 мм при температурі 200С,
. За цією формулою можна підрахувати, що в капілярі радіусом 0,01 мм при температурі 200С, 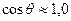 , висота підйому біля 1,5 м.
, висота підйому біля 1,5 м.

 ), а й поверхневого натягу (
), а й поверхневого натягу ( ):
): (5.15)
(5.15)
 а)
а)
 б)
б)
 .
. (5.16)
(5.16) - зміна поверхневого натягу зі зміною концентрації ПАР. Здатність ПАР до адсорбції визначається поверхневою активністю G – зміною поверхневого натягу при введенні перших порцій ПАР у розчині:
- зміна поверхневого натягу зі зміною концентрації ПАР. Здатність ПАР до адсорбції визначається поверхневою активністю G – зміною поверхневого натягу при введенні перших порцій ПАР у розчині: (5.17)
(5.17) , то адсорбція буде мати додатне значення (Г>0). Таку адсорбцію називають позитивною. Навпаки, коли поверхневий натяг збільшується із зростанням концентрації розчиненої речовини (
, то адсорбція буде мати додатне значення (Г>0). Таку адсорбцію називають позитивною. Навпаки, коли поверхневий натяг збільшується із зростанням концентрації розчиненої речовини ( ), то адсорбція буде мати від’ємне значення (Г<0). Таку адсорбцію називають негативною.
), то адсорбція буде мати від’ємне значення (Г<0). Таку адсорбцію називають негативною. а
а 



 для ряду водних розчинів насичених органічних кислот, співвідношення значень поверхневого натягу при однаковій концентрації для цього ряду приблизно однакове, тобто
для ряду водних розчинів насичених органічних кислот, співвідношення значень поверхневого натягу при однаковій концентрації для цього ряду приблизно однакове, тобто і т.д.
і т.д. 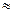 3,2.
3,2.
 (5.18)
(5.18) (5.19)
(5.19)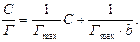 (5.20)
(5.20) , а тангенс кута нахилу прямої дорівнює
, а тангенс кута нахилу прямої дорівнює  .
.