
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Электрокинетические и электрокапиллярные явленияСодержание книги
Поиск на нашем сайте
Электрокинетические явления Электрокинетические явления отражают связь, существующую между относительным движением двух фаз (чаще всего жидкой и твёрдой) и электрическими свойствами границы их раздела. Электрокинетические явления возникают в тех случаях, когда одна фаза диспергирована в другой, то есть когда система может быть охарактеризована как микрогетерогенная. Различают четыре группы электрокинетических явлений: электроосмос, электрофорез, потенциал течения и потенциал осаждения.
Электрокинетические явления, особенно электроосмос и электрофорез, используются при обезвоживании и очистке различных материалов, нанесении на непроводящие материалы покрытий из каучука, отходов кожи и т.п., а также при пропитке тканей огнестойкими веществами, определении состава и разделении энзимов, белков, вирусов и других сложных систем и т.д. Само существование электрокинетических явлений указывает на то, что в месте контакта твёрдого тела и жидкости имеется двойной электрический слой, причём и твёрдое тело, и жидкость обладают определёнными зарядами. Изучение связи, существующей между направлением и скоростью электрофореза или электроосмоса, с одной стороны, и направлением и напряжённостью приложенного электрического поля – с другой, позволяет получить сведения о знаке и величине заряда твёрдых частиц относительно жидкости и о соответствующем ему скачке потенциала. Приближенная теория электрокинетических явлений приводит к следующим уравнениям для скорости электроосмоса U и потенциала течения Е: U = Е = где ee0 – диэлектрическая проницаемость жидкой фазы; DyD х – напряжённость электрического поля в направлении, параллельном границе раздела твёрдой и жидкой фаз; h – вязкость жидкой фазы; kо – её электропроводность; р – давление, вызывающее относительное перемещение фаз по границе раздела между ними; S – сечение взвешенной частицы (или поры). Величина z, входящая в уравнения, называется дзета-потенциалом или электрокинетическим потенциалом; его можно найти с помощью уравнений, если измерить U или Е. Уравнения в силу их приближённости позволяют определить лишь ориентировочные значения z-потенциала. Непосредственные измерения дают при этом не z, а eeоz /h, и расчётное значение z-потенциала зависит от выбора численных значений ee0 и h. Помимо того, напряжённость поля и удельная электропроводность жидкости, то есть величины, которые следовало бы использовать при расчёте z-потенциала по уравнениям, вблизи границы раздела и в глубине жидкой фазы в общем случае неодинаковы. Такое различие обусловлено тем, что у поверхности раздела фаз ионный состав обычно иной, чем средний ионный состав всей жидкой фазы. Считалось, что z-потенциал коллоидной химии – это то же самое, что электродный потенциал или E-потенциал электрохимии, точнее, та его часть, которая отвечает скачку потенциала на границе электрод – электролит. Для такого предположения имелись известные основания, вытекающие из самой природы электрокинетических явлений. Действительно, если граница раздела жидкость – твёрдое тело в то же время является плоскостью скольжения при относительном перемещении фаз, то z-потенциал должен быть равен E-потенциалу или, вернее, скачку потенциала gL,М . Если бы это предположение оказалось справедливым и отвечающим опыту, то появилась бы возможность экспериментального измерения скачка потенциала электрод – электролит, а следовательно, и определения абсолютного потенциала электрода. Имеется возможность косвенной проверки сопоставлением зависимости z-потенциала и gL,М-потенциала от состава раствора. Поскольку gL,М является единственной составной частью j-потенциала, зависящей от концентрации электролита, то при соблюдении равенства z = gL,М изменение z-потенциала и E-потенциала с концентрацией должно быть одинаковым и подчиняться одному и тому же закону. Результаты опытов показали, что отождествлять z- и E-потенциалы нельзя. E- и z-потенциалы изменяются с концентрацией по разным законам. При этом оказывается, что z-потенциал изменяется не монотонно, а проходит в отличие от E-потенциала через минимум или максимум. По своей абсолютной величине z-потенциал обычно меньше, чем E-потенциал, и стремится к нулю при повышении концентрации. В то время как знак E-потенциала сохраняется неизменным в широком интервале концентраций, знак z-потенциала может меняться на обратный и тем раньше, чем выше валентность ионов и чем сильнее их способность к избирательной адсорбции. Изменение знака z-потенциала (так называемая перезарядкаповерхности) приводит к тому, что для одной и той же поверхности раздела знак z-потенциала может быть или одинаковым, или обратным знаку E-потенциала. Таким образом, z-потенциал отличается по своей природе от E-потенциала. На основании свойств z-потенциала можно заключить, что он представляет собой некоторую часть той доли общего скачка потенциала, которая целиком расположена в жидкой фазе. Именно потому, что z-потенциал в отличие от E-потенциала лежит в одной и той же фазе, оказывается возможным прямое экспериментальное определение его абсолютной величины. Падение потенциала, соответствующее z-потенциалу, должно быть локализовано на границе между слоем жидкости, непосредственно примыкающем к поверхности твёрдого тела (и связанным с ним при относительном движении жидкой и твёрдой фаз), и более глубокими её слоями, удалёнными от поверхности раздела фаз. Существование между твёрдым телом и раствором наряду с общим скачком потенциала также z-потенциала следует учитывать при разработке теории строения двойного электрического слоя. Эта теория должна объяснить не только причины появления электрокинетического потенциала, но и характер его изменения с составом раствора и, в частности, явление перезарядки поверхности. Электрокапиллярные явления На границе раздела двух фаз можно выделить пограничный слой, так называемую поверхностную или пограничную фазу. Она обладает избытком свободной энергии по сравнению с каждой из граничащих фаз. Эта избыточная энергия, отнесённая к единице поверхности раздела фаз, то есть удельная свободная энергия s, имеет размерность Дж×м–2 или Н×м–1. В случае границы двух жидких фаз, например жидкого металла (ртути, амальгам, галлия) и раствора, удельная свободная энергия s совпадает с поверхностным или пограничным натяжением g, имеющим ту же размерность, что и s. Поверхностная фаза отличается от граничащих фаз не только энергетически, но и во многих других отношениях, в частности по составу. Содержание компонентов в поверхностной фазе может быть существенно иным, чем в объёме фаз. Подобное изменение состава является следствием взаимодействия частиц раствора с поверхностью твёрдого или жидкого металла и характеризует процесс адсорбции. В зависимости от природы сил взаимодействия различают физическую (электростатическую) адсорбцию, вызванную чисто кулоновским взаимодействием, специфическую, когда, например, ионы адсорбируются на одноимённо заряженной поверхности и обусловленную, в основном, силами Ван-дер-Ваальса, а также хемосорбцию, связанную с образованием химических соединений между адсорбентом (в данном случае металлом) и адсорбатом (в данном случае частицами, находящимися в растворе). Если концентрация частиц увеличивается по мере приближения к поверхности, то адсорбция называется положительной. Положительная адсорбция может быть обусловлена «выталкиванием» частиц из объема на поверхность. Так ведут себя гидрофобные органические вещества в водных растворах, которые нарушают водородные связи между молекулами воды в объеме раствора. Выигрыш энергии за счет восстановления этих связей при переходе органических молекул на поверхность и служит причиной их «выжимания» из объема раствора. Другой возможной причиной положительной адсорбции является взаимное притяжение между поверхностью электрода и частицами раствора. Это взаимодействие, как уже говорилось выше, может быть чисто электростатическим или специфическим. Если концентрация частиц убывает по мере приближения к поверхности, то адсорбция называется отрицательной. Отрицательная адсорбция частиц может быть вызвана или втягиванием сильно сольватированных частиц с поверхности в объем раствора, или отталкиванием ионов от одноименно заряженной поверхности электрода. Первый эффект приводит к заметной отрицательной адсорбции только в концентрированных растворах (1 моль/л), в то время как отталкивание ионов от одноименно заряженной поверхности электрода вызывает существенную отрицательную адсорбцию и в разбавленных растворах. Итак, в результате адсорбции в поверхностной фазе оказывается избыточное (или недостаточное) количество частиц определённого вида (или определённых видов), называемое обычно поверхностным избытком i -го компонента (или i -х компонентов) Г i . Он определяется как суммарное превышение концентрации данного компонента в поверхностной фазе над концентрацией того же компонента в объёме раствора, отнесённое к единице поверхности раздела, то есть имеет размерность моль×м–2. Величину Г i можно определить также как количество данного компонента i, которое нужно ввести в систему (или, наоборот, вывести из системы, если Г i < 0) для того, чтобы при увеличении поверхности раздела фаз на единицу состав объемных фаз остался без изменения. Как поверхностная энергия, так и поверхностные избытки зависят от разности потенциалов на границе раздела фаз. Следует различать понятия поверхностного избытка Г i и поверхностной концентрации А i, то есть количества компонента i, непосредственно связанного с единицей поверхности электрода. Обе величины (Г i и А i) имеют одинаковую размерность (моль/м2 ), однако поверхностная концентрация – величина всегда положительная, тогда как поверхностный избыток может быть как положительным, так и отрицательным. Электрокапиллярные явления отражают связь, существующую между поверхностным натяжением и разностью потенциалов на границе двух фаз. Графически эта связь выражается в виде электрокапиллярных кривых (э.к.к.). Впервые электрокапиллярные явления были изучены на границе ртути и водных растворов электролитов Липпманом (1875), который использовал для этой цели сконструированный им капиллярный электрометр. При снятии электрокапиллярных кривых на ртутный микроэлектрод, находящийся в капилляре и контактирующий с раствором, подаётся определённый потенциал и измеряется высота столба ртути, удерживаемого в стеклянной трубке над ртутным мениском в капилляре. Потенциал на границе между раствором и ртутью в капилляре задаётся наложением определённой ЭДС (например, от потенциометрической установки) на электрохимическую систему, в которой одним электродом служит капиллярный электрод, а другим – соответствующий электрод сравнения с известным значением потенциала. Как это следует из теории капиллярности, высота ртутного столба над ртутным мениском в капилляре является мерой поверхностной энергии на границе ртуть – раствор. Соотношение между этими двумя величинами можно записать в виде уравнения 2 p r s = p h r 2 g r, откуда s = где r – радиус капилляра; g – ускорение силы тяжести; r – плотность ртути. Измеряя высоту h при различных значениях потенциала и рассчитывая по уравнению соответствующие ей значения s, можно построить электрокапиллярную кривую. Для ряда разбавленных растворов (например, H2SO4, КОН, KNO3, Na2SO4 и др.) форма электрокапиллярной кривой почти не зависит от природы электролита и близка к параболе. Максимум на кривой наблюдается почти при одном и том же значении потенциала, лежащем между – 0,19 и – 0,21В по водородной шкале. Поверхностное натяжение в точке максимума также мало изменяется при переходе от одного из указанных растворов к другому и составляет 0,42-0,43 Дж×м–2. В то же время электрокапиллярные кривые, полученные в растворах других электролитов, а также в присутствии большинства органических неионизированных веществ, весьма заметно отличаются по своей форме от параболы. Они менее симметричны и их максимумы расположены при иных значениях E и s (см. рис. 19). Присутствие ионов брома, йода или серы смещает максимум электрокапиллярной кривой в сторону более отрицательных значений потенциалов и (особенно в области потенциалов, расположенных слева от точки максимума) уменьшает поверхностное натяжение. При достаточно отрицательных потенциалах эффект этих анионов исчезает и электрокапиллярные кривые сливаются друг с другом. Присутствие ионов таллия или ионов тетрабутиламмония сдвигает максимум электрокапиллярных кривых в сторону более положительных значений и уменьшает поверхностное натяжение; особенно заметно это при потенциалах более отрицательных, чем потенциал точки максимума. По достижении достаточно положительных потенциалов эффект этих катионов исчезает. Присутствие амилового спирта изменяет форму электрокапиллярной кривой главным образом в области потенциалов, примыкающих к потенциалу максимума. С удалением в обе стороны от потенциала электрокапиллярного максимума эффект добавки амилового спирта снижается и кривая, снятая с добавкой спирта, совпадает с кривой для чистого раствора. Ионные или молекулярные соединения, оказывающие подобное действие, называются специфически адсорбирующимися или поверхностно-активными ионами или молекулами.
Электрокапиллярные свойства границы ртуть – раствор электролита можно объяснить, если допустить, что в отсутствие внешней ЭДС ртуть при потенциале E1 оказывается заряженной положительно по отношению к раствору. Поэтому со стороны раствора у границы раздела будет избыток отрицательных ионов. Присутствие одноимённо (положительно) заряженных ионов ртути на поверхности металла неизбежно приводит к появлению отталкивающих сил, и поверхностное натяжение на границе ртуть – раствор не может быть высоким; оно отвечает некоторой величине s1 (см. рис. 20). При наложении внешней ЭДС потенциал ртути смещается вследствие её катодной поляризации в сторону более отрицательных значений, например, до величины E2 . Часть ионов ртути при этом нейтрализуется электронами, и её заряд, оставаясь положительным, соответственно снижается. Это уменьшает силы отталкивания между поверхностными ионами ртути, а следовательно, повышает поверхностное натяжение, например, до величины s2 ; одновременно уменьшается число отрицательных ионов, притянутых к поверхности ртути со стороны раствора. При определённом значении потенциала E = Eq=0 положительные заряды всех ионов ртути будут скомпенсированы электронами и заряд поверхности ртути станет равным нулю; одновременно (если в растворе нет поверхностно-активных частиц) заряд раствора также станет равным нулю и поверхностное натяжение достигает максимального значения smax . При дальнейшем смещении потенциала ртути в отрицательную сторону до величины E3 на её поверхности появляются избыточные электроны, к которым со стороны раствора притягиваются положительно заряженные ионы. Снова возникают силы отталкивания между одноимённо заряженными, но теперь уже отрицательными частицами, и поверхностное натяжение упадёт до некоторой величины s3. По мере увеличения отрицательного значения потенциала избыточный отрицательный заряд ртути будет увеличиваться, а поверхностное натяжение уменьшаться. Восходящая ветвь электрокапиллярной кривой относится, таким образом, к положительно, а нисходящая – к отрицательно заряженной поверхности ртути.
Если исходить из предположения, что адсорбция ионов на ртути определяется исключительно электростатическими силами, то все анионы должны изменять ход лишь восходящей ветви электрокапиллярной кривой, где поверхность ртути заряжена положительно. Напротив, влияние катионов должно локализоваться только на нисходящей ветви, где они электростатически притягиваются к отрицательно заряженной поверхности ртути. В действительности, как это было найдено ещё Гуи, многие анионы изменяют ход электрокапиллярной кривой справа от точки максимума, а некоторые катионы влияют не только на нисходящую, но и на восходящую ветвь кривой. Такое поведение ионов нельзя объяснить действием только кулоновских сил. Оно связано с силами взаимодействия, отличными от простых электростатических сил. Такими силами, специфическими для данного рода частиц, могут быть, например, силы Ван-дер-Ваальса или химические (валентные). Благодаря этим силам ионы в состоянии удерживаться на одноимённо заряженной поверхности ртути. Точно так же нельзя на основе одних только электростатических представлений объяснить влияние неионизированных органических веществ на ход электрокапиллярных кривых. Большинство органических веществ обладает меньшей диэлектрической постоянной, чем вода, и поэтому должны были бы изгоняться ею из двойного слоя уже при незначительных зарядах подобно тому, как диэлектрик с меньшей диэлектрической постоянной вытесняется из заряженного конденсатора диэлектриком с большей диэлектрической постоянной. В этом случае эффект органических веществ должен был бы проявляться только в очень узкой области потенциалов, непосредственно примыкающих к максимуму, электрокапиллярной кривой. В действительности, изменение формы электрокапиллярной кривой под действием органических веществ наблюдается даже при потенциалах, удалённых на 0,5-0,8 В в обе стороны от потенциала максимума электрокапиллярной кривой, то есть здесь должны проявляться какие-то специфические силы.
Лекция 11
|
|||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-08-26; просмотров: 1187; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.137.186.26 (0.01 с.) |

 ,
, или Е =
или Е =  ,
, r g r,
r g r,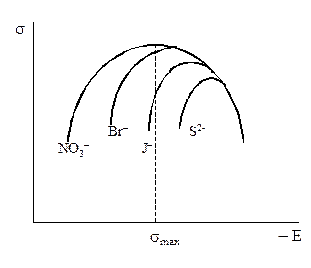 а
а
 б
б
 в
в




