Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Хромосомные болезни человекаСодержание книги
Поиск на нашем сайте
Хромосомными болезнями (хромосомными синдромами) называют комплексы множественных врожденных пороков развития, вызываемых численными (геномные мутации) или структурными (хромосомные аберрации) изменениями хромосом, видимыми в световой микроскоп. Хромосомные аберрации и изменения количества хромосом могут возникать на разных этапах развития организма. Если они происходят в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант). Если аномалия возникает в процессе эмбрионального развития при дроблении зиготы, кариотип плода будет мозаичным. Формируется такая мозаика следующим образом. В норме все бластомеры содержат одинаковый набор хромосом, идентичный набору зиготы. Однако в отдельных случаях возможно нарушение расхождения хромосом, вследствие чего в разные бластомеры попадет неодинаковое их количество (один бластомер с моносомией, а другой — с трисомией). При последующем дроблении возникают две клеточные линии — клоны, сохраняющие особенности аномального кариотипа. В зависимости от стадии, на которой произошло нарушение, и интенсивности размножения клеток число этих клеточных линий может быть различным. Клетки, берущие начало от нормальных бластомеров, будут иметь неизмененный кариотип. Такое явление называется генетическим мозаицизмом. Мозаичные организмы могут содержать несколько (2, 3, 4 и более) клеточных клонов с различными кариотипами. Это явление иногда сопровождается мозаицизмом во всех либо в отдельных органах и системах. При незначительном количестве аномальных клеток фенотипические проявления могут не обнаруживаться. Геномные мутации у человека представлены триплоидией и анеуплоидиями. Нарушение плоидности представлено лишь одним заболеванием — синдромом триплоидии. Это летальная мутация — ребенок умирает до рождения или в первые часы после рождения. Синдромы трисомий — наиболее частая форма численных изменений хромосом. Полная моносомия, совместимая с жизнью, наблюдается только по Х-хромосоме. В основе синдромов, обусловленных структурными нарушениями хромосом, лежат либо частчастичные трисомии (при дупликациях и транслокациях), либо частичные моносомии (при делециях), либо их сочетания. Хромосомные болезни у новорожденных детей встречаются с частотой примерно 2, 4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомий по крупным хромосомам, моносомии) несовместимы с жизнью — эмбрионы и плоды элиминируются из организма матери в основном в ранние сроки беременности. Окончательный диагноз хромосомных болезней ставится с помощью цитогенетических методов.
Трисомиия
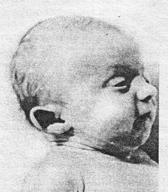
Рис. 11.1. Больной с синдромом Патау (трисомия 13)
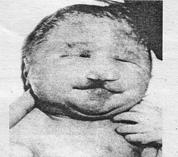
Наиболее часто у человека встречаются трисомии по 13-й, 18-й и 21-й парам хромосом. Синдром Патау (синдром трисомии 13) встречается с частотой 1: 6000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Дети с синдромом Патау (рис. 11.1) рождаются с массой тела ниже нормы (2500 г). У них наблюдаются умеренная микроцефалия, нарушение развития различных отделов центральной нервной системы, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80% новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое. Большинство больных с синдромом Патау (98%) умирают в возрасте до года, оставшиеся в живых страдают глубокой - идиотией. Синдром Эдвардса (синдром трисомии 18) встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще рождаются у женщин после 35 лет. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7%. Цитогенетически синдром Эдвардса представлен простой трисомией 18. У девочек он встречается значительно чаще, чем у мальчиков, что связано, возможно, с большей жизнестойкостью женского организма. Дети с трисомией 18 рождаются с низкой массой тела (в среднем 2177 г), хотя сроки беременности нормальные или даже превышают норму. Фенотипические проявления синдрома Эдвардса многообразны (рис. 11.2).
Рис. 11.2. Больной с синдромом Эдвардса (трисомия 18)
Наиболее часто отмечаются аномалии мозгового и лицевого черепа. Мозговой череп долихоцефалической формы. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка уха, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80% случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив. Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают в возрасте до 3 месяцев; до года доживает лишь один ребенок из десяти; оставшиеся в живых — глубокие олигофрены. Синдром Дауна (синдром трисомии 21) — наиболее часто встречающаяся форма хромосомной патологии у человека — 1:750. Достоверно установлено, что дети с синдромом Дауна чаще рождаются у немолодых родителей. Если возраст матери 41-46 лет, то вероятность рождения больного ребенка возрастает до 4,1 %.
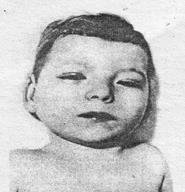
Цитогенетически синдром Дауна представлен простой трисомией (94% случаев), транслокационной формой или мозаицизмом (4% и 2% соответственно). За возникновение фенотипических проявлений синдрома Дауна отвечает лишь небольшой участок длинного плеча 21-й хромосомы (21q22.3) и независимо от механизма удвоения развивается типичная клиническая картина. Масса тела новорожденных с синдромом Дауна в среднем 3167 г. Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея (рис. 11.3).
Рис. 11.3. Больной с синдромом Дауна (трисомия 21)
Из пороков внутренних органов наиболее типичны дефекты сердечно-сосудистой системы (межжелудочковой или межпредсердной перегородок и др.) и органов пищеварения (атрезии и стенозы различных отделов), У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживаются катаракты. Характерна умственная отсталость, преимущественно имбецильность (65-90%); дебильность и идиотия диагностируются примерно в равном соотношении. Средняя продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции.
Частичные трисомии Помимо полных трисомии известны синдромы, связанные с частичными трисомиями практически по любой хромосоме. Однако они встречаются реже одного случая на 100 000 рождений. Синдром трисомии по короткому плечу 9-й хромосомы (9р+) — наиболее часто встречающаяся форма частичных трисомии. Для больных с трисомией 9р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные уголки рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитые ногти и дистальные фаланги пальцев рук. Часто наблюдаются выступающие лобные кости,.повышенная обволошенность, пятна цвета "кофе с молоком" на коже, эпикант, косоглазие, высокое дугообразное нёбо, короткая шея, сколиоз, частичная синдактилия пальцев стоп. Примерно в четверти случаев обнаруживаются врожденные пороки сердца. Прогноз для жизни сравнительно благоприятный — описаны больные, достигшие преклонного возраста.
11.3. Частичные моносомии
Синдромы частичных моносомии встречаются примерно с такой же частотой, как и синдромы частичных трисомии. Наиболее известные из них синдромы Вольфа-Хиршхорна и "кошачьего крика". Синдром Вольфа -Хиршхорна (4р-) обусловлен делецией короткого плеча 4-й хромосомы. Популяционная частота заболевания — примерно 1 случай на 100 000. Дети с синдромом Вольфа—Хиршхорна-обычно рождаются у молодых родителей, доношенными, но со значительно сниженной массой тела (около 2000 г). Для таких детей характерна резкая задержка физического и психомоторного развития. У них наблюдаются умеренно выраженная микроцефалия, клювовидный нос, выступающее надпереносье, деформированные низко расположенные ушные раковины, вертикальные складки кожи впереди ушных раковин, гипотония мышц, значительное снижение реакции на внешние раздражения, судорожные припадки. Отмечаются также расщелины верхней губы и нёба, деформации стоп, аномалии глазных яблок, эпикант и маленький рот с опущенными уголками. Из внутренних органов чаще поражается сердце (пороки развития), примерно в половине случаев — почки (гипоплазия и кисты). Большинство детей с синдромом 4р- умирают на первом году жизни. Максимально известный возраст пациента с этим синдромом — 25 лет. Синдром "кошачьего крика" (5р-) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома — примерно 1: 45 000. Для данного синдрома наиболее характерны специфический плач, напоминающий кошачье мяуканье, лунообразное лицо, мышечная гипотония, умственное и физическое недоразвитие, микроцефалия, низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей, косоглазие (рис. 11. 4). Иногда наблюдаются аномалии глаз (недоразвитие зрительного нерва, очаги депигментации сетчатки). Наиболее постоянный признак синдрома — "кошачий крик" — обусловлен изменениями гортани: сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой, уменьшением надгортанника. Изменения других органов и систем неспецифичны. Продолжительность жизни значительно снижена, только около 14% больных переживают возраст 10 лет.
Рис. 11.4. Больной с синдромом кошачьего крика" (5р.)
Синдром Орбели (13q-) обусловлен делецией длинного плеча 13-й хромосомы сегментов 13q22-q31. Популяционная частота синдрома не установлена. Дети с синдромом Орбели рождаются c низкой массой тела (2200 г). Клинически синдром сопровождается аномалиями развития всех систем органов. Характерны микроцефалия, отсутствие носовой вырезки (лоб непосредственно переходит d нос), эпикант, антимонголоидный разрез глаз, широкая г пинка носа, высокое нёбо, низко расположенные деформированные ушные раковины. Отмечаются поражения глаз (микрофтальмия, иногда анофтальмия, косоглазие, катаракта, рстинобластома), опорно-двигательного аппарата (короткая шея, гипо- или аплазия первого пальца кисти и пяточной кости, синдактилии кистей и стоп), атрезии прямой кишки и заднепроходного отверстия. Часты пороки развития сердца, почек, головного мозга. Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потери сознания, судороги. Большинство больных с синдромом I3q- погибают на первом году жизни.
ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ И БОЛЕЗНИ С НАСЛЕДСТВЕННОЙ ПРЕДРАСПОЛОЖЕННОСТЬЮ Как уже отмечалось, врожденные пороки развития — это стойкие морфологические изменения тканей или органов, выходящие за пределы вариаций их строения. Формирование врожденных пороков развития в результате нарушения нормального течения эмбриогенеза называется тератогенезом (от греч. teras — урод, чудовище). Наука об этиологии, патогенезе и проявлениях врожденных пороков развития называется тератологией. Частота грубых врожденных пороков развития, сопровождающихся нарушением функций, в популяциях человека составляет 2-3%. Формирование врожденных пороков — результат отклонений от нормального развития особи. Единый процесс онтогенеза состоит из следующих этапов: 1) гаметогенез; 2) оплодотворение; 3) эмбриональный морфогенез; 4) постэмбриональное развитие. В результате гаметогенеза образуются половые клетки, несущие в себе генетическую информацию, в процессе реализации которой из одной клетки (зиготы) развивается многоклеточный организм. При оплодотворении происходит объединение генетической информации материнского и отцовского организмов, что обусловливает комбинативную изменчивость. Формирование морфологических структур эмбриона — эмбриональный морфогенез — включает эмбриональный гистогенез (возникновение специализированных тканей из слабо дифференцированных клеток эмбриональных зачатков) иорганогенез (развитие органов и систем органов). Эмбриональный морфогенез осуществляется при взаимодействии генотипа зародыша и организма матери и связан с процессами размножения, роста, дифференцировки, миграции и отмирания клеток. Эти процессы контролируются сложными взаимодействиями генетических, эпигеномных и внешних факторов, определяющих в конечном итоге временную и пространственную последовательность экспрессии (включения и выключения) генов и тем самым дифференцировку клеток и морфогенез. Нарушение в процессе эмбриогенеза любого из вышеперечисленных механизмов вызывает отклонение от нормального развития, что может проявиться в виде врожденного порока. В постэмбриональный период также возможно проявление некоторых пороков развития. На внутриклеточном уровне к "пусковым" механизмам нарушения развития относятся изменения молекулярных процессов репликации ДНК, биосинтеза белков-ферментов и др. К основным клеточным механизмам тератогенеза относятся нарушения размножения, миграции и дифференцировки клеток. Результатом снижения митотической активности клеток могут быть гипоплазии или аплазии органа или его части. В результате нарушения миграции клеток могут развиваться гетеротопии и другие пороки. Дифференцировка, т. е. образование разнородных клеток, тканей и органов из однородного эмбрионального зачатка, происходит последовательно в течение всего эмбриогенеза. Основной механизм специализации клеток — дифференциальная активность генов, в результате которой в разные фазы эмбриогенеза синтезируются специфические для каждой стадии ферменты, обеспечивающие специализацию клеток. Нарушения механизмов включения и выключения отдельных блоков генов приводит к развитию различных пороков. К основным механизмам тератогенеза на тканевом уровне относятся постепенное отмирание отдельных клеточных масс, замедление распада и рассасывания клеток, отмирающих в ходе нормального эмбриогенеза, и нарушение адгезии тканей. Физиологическая гибель клеток происходит под действием лизосомальных ферментов в процессе окончательного формирования органов. Такая первичная гибель клеток наблюдается при слиянии нёбных отростков, открытии естественных отверстий, формировании пальцев. Задержка или замедление физиологического распада клеток может приводить к синдактилии, атрезиям и другим порокам развития.
|
||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 198; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.119 (0.011 с.) |