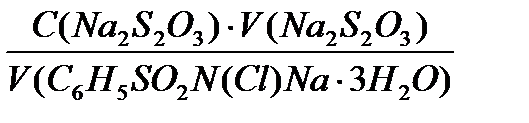Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Объемный анализ (титриметрия)Содержание книги Похожие статьи вашей тематики
Поиск на нашем сайте
ХИМИЧЕСКИЕ МЕТОДЫ
Эти методы основаны на выполнении некоторых химических реакций между исследуемым веществом и реактивами, в результате чего можно произвести измерение количества определяемого вещества. Основными химическими методами являются весовой (гравиметрия) и объемный (титриметрия). Весовой метод Определяемый элемент в навеске анализируемого вещества при помощи химической реакции переводят в осадок. Полученный осадок отфильтровывают, промывают, высушивают (если необходимо, прокаливают) и взвешивают. Зная массу исходного вещества, массу и состав полученного осадка, можно рассчитать содержание определяемого элемента в исследуемом веществе. К достоинствам этого метода относятся: а) большая универсальность, т.к. с его помощью можно определить количество практически любого элемента, и б) большая точность определения, которая достигается применением аналитических весов(вследствие этого возможные ошибки не превышают 0,2%).Но этот метод имеет большой недостаток- гравиметрические определения требуют затрат большого количества времени и труда. Например, содержание сульфат-ионов в растворе можно определить по массе осадка сульфата бария, образующегося в ходе реакции: Ва+2 + SO42- = BaSO4¯ Объемный анализ (титриметрия) В основе объемного метода лежит титрование – к раствору одного вещества приливают раствор другого вещества, пока они не прореагируют нацело. Точно измеряя объемы смешиваемых растворов и зная концентрацию одного из них, можно на основании закона эквивалентов рассчитать концентрацию другого из них. Один из растворов содержит вещество неизвестной концентрации и представляет собой анализируемый раствор. Второй раствор, который содержит реагент, концентрация которого известна с большой точностью, называется рабочим раствором или титрантом. Момент в процессе титрования, когда количества реагирующих веществ в смеси становятся эквивалентными (прореагируют нацело) называется моментом эквивалентности или эквивалентной точкой. Достоинством этого метода является быстрота определения при достаточной его точности. Недостаток метода - трудность определения конца титрования и некоторые другие требования к реакциям, лежащим в основе объемных определений, меньшая точность (измерение объемов производится с меньшей точностью, чем взвешивание на аналитических весах). К химическим реакциям, используемым в объемных методах количественного анализа, предъявляются следующие основные требования: а) Наличие способа фиксирования точки эквивалентности. б) Достаточно большая скорость реакции. Вещество, содержащееся в одной капле добавленного раствора, должно вступить в реакцию за время, необходимое для перемешивания этой капли с остальным раствором. Если реакция протекает медленно, то невозможно определить момент эквивалентности. в) Практическая необратимость (большая величина константы равновесия). г) Отсутствие побочных реакций. МЕТОД НЕЙТРАЛИЗАЦИИ С помощью метода нейтрализации определяют кислоты (алкалиметрия) и основания (ацидиметрия). Реакциями нейтрализации в водных растворах являются все реакции между кислотами и основаниями, один из продуктов которых является вода. В соответствии с протолитической теорией Бренстеда-Лоури кислотой является любое вещество-донор протона, а основанием- акцептор протона. Кислота и получившееся при отдаче протона основание составляют сопряженную пару. Таким образом, реакция нейтрализации согласно протолитической теории представляет собой протолитическое равновесие: НА + В = ВН+ + А- кислота 1 основание 2 кислота 2 основание 1 Одним из компонентов протолитической реакции является растворитель. С точки зрения кислотно-основных свойств растворители можно разделить на три группы: 1. Апротонные растворители, не обладающие ни кислотными, ни основными свойствами (например, бензол, гексан, хлороформ). 2. Протофильные растворители, обладающие только основными свойствами (например, ацетон, диоксан, диэтиловый эфир, пиридин). 3 .Амфипротные растворители, обладающие как кислотными, так и основными свойствами(вода, спирты, карбоновые кислоты, первичные и вторичные амины). Важнейшая особенность амфипротных растворителей - способность к передаче протона от одной молекулы к другой. Такие процессы называются автопротолизом. Например, для воды: Н2О + Н2О = Н3О+ + ОН- для аммиака NH3 + NH3 = NH4+ + NH2- для муравьиной кислоты НСООН + НСООН = НСОО- + НСООН2+ Характеристикой равновесия автопротолиза служит константа автопротолиза. Для воды константа автопротолиза обозначается Кw и называется ионным произведением воды: Kw = ан3о+ ·аон- = 1,0 · 10-14 (25оС). Величина, равная –lgан3о+ обозначается рН. Если ан3о+ = аон- рН = ½ рКw среда нейтральная ан3о+ > аон- рН < ½ рКw среда кислая ан3о+ < аон- рН = ½ рКw среда щелочная При взаимодействии с амфипротными растворителями растворенные вещества могут проявлять как кислотные, так и основные свойства: CH3COO- + H2O = OH- + CH3COOH NH3 + H2O = NH4+ + OH- В неводных растворителях: NH3 + HCOOHбезв. = NH4+ HCOO- NH4+ + CH3OHбезв. = CH3OH2+ + NH3
Простейшей реакцией нейтрализации является взаимодействие сильной кислоты со щелочью в водном растворе: Н3О+ + ОН- = 2Н2О Реакция между ионами гидроксония и гидроксила сильно смещена вправо в соответствии с малой величиной ионного произведения воды. Таким образом, эта реакция является практически необратимой. Кроме того, реакция переноса протона является очень быстрой. Поэтому в эквивалентной точке ан3о+ = аон- и рН=7 Если в реакции нейтрализации участвует слабая кислота, например, уксусная кислота (СН3СООН + ОН- = СН3СОО- + Н2О), то реакция нейтрализации заметно обратима. В эквивалентной точке реакция среды отличается от нейтральной и вследствие наличия в равновесной смеси группы ОН- рН >7. В объемном анализе применяется также взаимодействие сильной кислоты и слабого основания. Слабыми основаниями являются аммиак, амины, анионы слабых кислот, входящие в состав солей. Примеры таких реакций: NH3 + H3O+ = NH4+ + H2O CO32- + H3O+ = HCO3- + H2O Реакция нейтрализации слабого основания является заметно обратимой, и в момент эквивалентности имеются ионы Н3О+. Следовательно, в точке эквивалентности в этом случае рН <7. Таким образом, используя метод нейтрализации, можно определять количество кислот, оснований и других веществ, реагирующих с кислотами и основаниями. Используя ионообменники, можно также определять вещества, не реагирующие с кислотами и основаниями (например, NaCl, Na2SO4.и др.). Порядок выполнения работы
1. Бюретку заполняют раствором NaOH, полученным у преподавателя. 2. Пипетку споласкивают два раза раствором щавелевой кислоты. 3. В колбу для титрования переносят пипеткой 3(или 5) мл щавелевой кислоты и добавляют 1 каплю фенолфталеина. 4. Подносят колбу к бюретке с таким расчетом, чтобы носик бюретки входил в горлышко колбы, не касаясь стенок. Одной рукой совершают вращательные движения колбы, а другой рукой оттягивают резинку у бусинки (шарика) бюретки, спуская по каплям раствор из бюретки. Необходимо внимательно следить за окраской раствора. Титрование заканчивают, когда раствор принимает от очередной капли щелочи слабо-розовую окраску, не исчезающую в течение 30 секунд. 5. В лабораторный журнал записывают объем щелочи, пошедший на титрование кислоты с точностью до двух сотых миллилитра. Промывают колбу дистиллированной водой, снова заполняют бюретку, устанавливают на нуль и подобным образом проводят еще два повторных титрования.
Составляют таблицу.
V (NaOH) = (V1 + V2 + V3) / 3
Расчет: С (NaOH) = C (1/2 H2C2O4) ·V(Н2С2О4) / V(NaOH)ср.
(¦экв (H2C2O4) = 1/2; C (1/2 H2C2O4) = 0,1 моль/л) МЕТОД ПЕРМАНГАНАТОМЕТРИИ Метод основан на реакции MnO4- + 8H+ + 5e = Mn2+ +4H2O φo = 1,51 В Перманганат калия обладает высокой окислительной способностью, он дешев, растворы KMnO4 обладают достаточной устойчивостью при правильном хранении - этим объясняется широкое применение КMпО4, в объемном анализе. Окисление раствором КMпО4 может проходить в кислой среде, нейтральной или щелочной. Однако количественные определения по методу перманганатометрии чаще всего проводят в кислой среде по следующим причинам: 1) в кислой среде MnO4- переходит в Mn2+, проявляя наибольшую окислительную активность; 2) в кислой среде все продукты реакции бесцветны и растворимы. В нейтральной или слабо щелочной среде МnO4- восстанавливается в труднорастворимый оксид МnO2, что затрудняет определение эквивалентной точки. Для подкисления титруемых растворов используется серная кислота. Азотную кислоту брать нельзя, т.к. она сама - сильный окислитель и может вступать в реакции с определяемыми восстановителями. Соляная кислота - восстановитель и может вступать в реакции с KMnO4. Специального индикатора для определения точки эквивалентности в перманганатометрии не требуется, т.к. растворы, содержащие ионы MnO4-,интенсивно окрашены и первая избыточная капля его окрашивает титруемый раствор в розовый цвет. По методу перманганатометрии можно определять: а) прямым титрованием - количество восстановителей – Fe2+, H2O2, щавелевой кислоты и ее солей, нитритов и т.д.; б) титрованием по замещению - количество веществ, реагирующих с восстановителями, например Са2+, который реагирует с C2О42-. Для выполнения определения осаждают Са2+ в форме СаС2.О4, осадок отделяют фильтрованием, растворяют в 2н H2SО4 и выделившуюся H2C2O4, титруют раствором КмпО4; в) титрованием по остатку - количество веществ, реагирующих с восстановителями, например K2Cr2O7, который реагирует с солью Мора (NH4)2Fe(SO4)2 · 6H2O - избыток соли Мора титруют раствором КMnО4. Метод перманганатометрии широко применяется в клинической лаборатории, а также при изучении физической химии, биохимии и гигиены. Например, определение количества кальция в крови, определение каталазы в крови по Баху и Зубковой, определение окисляемости воды и т.д. выполняют перманганатометрическим методом. Приготовление рабочего раствора КMпО4 Рабочий раствор КМпО4 относится к растворам с установленной концентрацией. Свежеприготовленный раствор KMnO4 некоторое время меняет свою концентрацию за счет небольших загрязнений, имеющихся в воздухе и попадающих в раствор (пыль органического происхождения, NH3, бактерии и т.п.). Так как количество примесей невелико, то и скорость окисления мала, и процесс заканчивается через 7-10 дней. После этого срока концентрация раствора КМпО4 становится постоянной и можно приступать к ее определению. .Приготовление раствора перманганата калия ведут следующим образом: 1. Рассчитывают необходимую навеску, исходя из необходимой концентрации и объема раствора: fэкв(KMnO4) = 1/5. m(KMnO4) = C(1/5 KMnO4) M(KMnO4) · 1/5 · V. Например, для приготовления 500 мл 0,02н KMnO4 m(KMnO4) = 0,02 · 158,04 · 1/5 · 0,5 = 0,3161 г 2. Берут навеску KMnO4 на техно-химических весах на 10% больше рассчитанной, т.к. часть его будет израсходована на окисление примесей, и растворяют в нужном объеме свежеперегнанной дистиллированной воды. 3. Приготовленный раствор хранят в темноте 7-10 дней плотно закупоренным, т.к. свет и присутствие СО2 способствуют разложению KMnO4: 4 KMnO4 + 4CO2 + 2H2O = 4KHCO3 + 4MnO2 + 3O2 4. Выпавший осадок МпО2 отфильтровывают через стеклянный фильтр, стеклянную вату или асбест (но не через бумажный фильтр). Оставлять МпО2 в растворе нельзя, т.к. он каталитически ускоряет процесс саморазложения КМпО4, а также участвует в реакциях окисления-восстановления, что снижает точность титрования. 5. Отфильтрованный раствор сохраняют в темных бутылях, защищая от пыли и прямого солнечного света. В качестве исходных веществ для определения концентрации раствора KMnO4 применяют кристаллическую щавелевую кислоту H2C2O4 · 2H2O или ее натриевую соль Na2C2O4. Оба эти вещества удовлетворяют требованиям, предъявляемым к исходным веществам, но удобнее пользоваться оксалатом натрия, т.к. он не содержит кристаллизационной воды и негигроскопичен. Лабораторная работа ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ ~ 0,02н KMnO4 по Na2C2O4. В основе определения концентрации рабочего раствора KMnO4 по оксалату натрия лежит реакция: 2KMnO4 + 5Na2C2O4 + 8H2SO4 = 2MnSO4 + 5Na2SO4 + K2SO4 + 10CO2 + 8H2O Приготовление 100 мл 0.02 н раствора оксалата натрия (исходное вещество) по точной навеске а) Расчет навески m(Na2C2O4) = C(1/2 Na2C2O4) · M(Na2C2O4) · 1/2· Vколбы = 0,02· 67,00· 0,1 = 0,134 г. б) Взвешивание и растворение Оксалат натрия отвешивает на аналитических весах с точностью до 0,0002 г, причем не нужно стремиться взвесить точно 0,134 г оксалата натрия. Важно, чтобы взятая навеска была правильно взвешена. Вполне допустимо отклонение величины взятой навески от рассчитанной на 10%. Практически поступают так: 1) Рассчитывают минимальную и максимальную навески: m min= 0,134 - 0,0134 = 0,1206 г. m max = 0,134 + 0.0134 = 0,1474 г. 2) Взвешивают на аналитических весах бюкс. Например, масса.бюкса mб = 20,3715 г. 3) Прибавляют к массе бюкса минимальную и максимальную навески mб.+ mmin.= 20,3715 + 0,1206 = 20,49021 г mб.+ mmax. = 20,3715 + 0,1474 = 20,5189 г Так определяются пределы взвешивания. 4) Взятую навеску количественно переносят в мерную колбу на 100 мл: оксалат натрия осторожно пересыпают через сухую воронку в колбу, осторожно промывают воронку маленькими порциями дистиллированной воды и снимают ее. 5) Взвешивают на аналитических весах бюкс с остатками соли и рассчитывают размер взятой навески по разности между массой бюкса с навеской и массой пустого бюкса(после пересыпания соли), например: m = 20,5048 – 20,3715 = 0,1333 г. в) Расчет концентрации C(1/2 Na2C2O4) = m(навески) / M(1/2 Na2C2O4) V(колбы) Например, c(1/2 Na2C2O4) = 0,1333 / 67,00· 0,1 = 0, 0199 моль/л Порядок титрования В бюретке - раствор KMnO4, в колбе для титрования -раствор оксалата натрия и серная кислота. Ход определения 3 мл раствора оксалата натрия переносят пипеткой в колбу для титрования, добавляют 2-3 мл 2н раствора Н2SО4, нагревают смесь до 80o С (для ускорения реакции), не доводя до кипения (во избежание разложения оксалата), и титруют раствором перманганата калия из бюретки. Реакция между Na2C2O4 и КMпО4 идет сначала очень медленно. Реакция идет ступенчато, причем сам перманганат с оксалатом, по существу, не реагирует. Стадии реакции: 1) 3MnSO4 + 2KMnO4 + 2H2O = 5MnO2 + K2SO4 + 2H2SO4 2) MnO2 + H2C2O4 + H2SO4 = MnSO4 + 2CO2 + 2H2O В первоначально взятом растворе KMnO4 имеются только следы Мп2+, поэтому обе реакции идут очень медленно. По мере накопления ионов Mn2+ как продукта реакции скорость реакции возрастает. Ионы Mn2+ являются катализатором данной реакции. Так как катализатор - продукт реакции, то реакция между оксалатом и перманганатом - пример автокаталитической реакции. Титрование выполняют следующим образом: расгвор KMnO4 приливают медленно, по каплям, тщательно перемешивая содержимое колбы и добавляя следующую каплю только после того, как обесцветилась предыдущая. Затем можно титровать быстрее, причем, если раствор остыл, его подогревают еще раз. Таким образом, условия титрования оксалата перманганатомтаковы: I) рН < 7 2) Нагревание до 70 -80°C. 3) Медленный (постепенно возрастающий) темп титрования. Титрование заканчивают, когда от очередной капли KMnO4 раствор в титровальной колбе станет блендно0розовым. Лабораторная работа ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ПЕРОКСИДА ВОДОРОДА Н2О2 В основе определения лежит реакция: 2KMnO4 + 5H2O2 + 3H2SO4 = 2MnSO4 + K2SO4 + 5O2 + 8H2O f(KMnO4) = 1/5; f(H2O2) = 1/2. Индикатор – первая избыточная капля перманганата калия. Порядок титрования В бюретке – раствор перманганата калия, в колбе для титрования – раствор пероксида водорода и 2н раствор серной кислоты. Ход определения Полученный раствор пероксида водорода доводят дистиллированной водой до метки и тщательно перемешивают. 3 мл раствора пипеткой переносят в колбу для титрования, добавляют 2-3млл 2н раствора серной кислоты и титруют рабочим раствором перманганата калия. Титрование выполняют при комнатной температуре. Расчет V(KMnO4) · C(1/5 KMnO4) = V(H2O2) · C(1/2 H2O2) C(1/2 H2O2) = V(KMnO4) · C(1/5 KMnO4) / V(H2O2) m(H2O2) = C(1/2 H2O2) ·V(колбы) · M(1/2 H2O2) Лабораторная работа ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ДИХРОМАТА КАЛИЯ Перманганатометрическое определение окислителей выполняется методом обратного титрования (или титрованием по остатку). Определение дихромата калия основано на последовательном протекании двух реакций: 1) K2Cr2O7 + 6FeSO4 + +7H2SO4 = K2SO4 + 3Fe2(SO4)3 + Cr2(SO4)3 + 7H2O f(K2Cr2O7) = 1/6; f(FeSO4) = 1. 2) 10FeSO4 + 2KMnO4 + 8H2SO4 = 5Fe2(SO4)3 + K2SO4 + 2MnSO4 + 8H2O f(KMnO4) = 1/5; f(FeSO4) = 1. Определение протекает в две стадии: 1) К определенному объему раствора дихромата калия прибавляют точно измеренный объем рабочего раствора соли Мора (NH4)2Fe(SO4)2 · 6H2O, причем соль Мора берут в избытке. 2) Остаток соли Мора оттитровывают рабочим раствором КМпО4. Индикатор - первая избыточная капля КМпО4. Приготовление рабочего раствора соли Мора (NH4)2Fe(SO4)2 · 6H2O Длительное хранение рабочего раствора соли Мора невозможно из-за окисления Fe2+ кислородом воздуха. Поэтому раствор готовят непосредственно перед применением. Процесс окисления тормозится при добавлении больших количеств кислоты, которая также предотвращает гидролиз соли. Расчет навески m(соли Мора) = C(FeSO4)· M((NH4)2Fe(SO4)2·6H2O)· V(колбы) Например, необходимо приготовить 100 мл 0,02н раствора соли Мора: m(соли Мора) = 0,02 · 392,14 · 0,1 = 0,7843 г 0,7-0,9 г соли Мора взвешивают на технохимических весах, пересыпают в склянку на 100 мл, куда предварительно налито 50мл 2н H2SO4, перемешивают до растворения соли, добавляют 50мл дистиллированной воды и тщательно перемешивают раствор. Раствор соли Мора является вспомогательным, его концентрацию не необходимости рассчитывать. Порядок титрования В бюретке - раствор перманганата калия, в колбе для титрования - раствор соли Мора или смесь раствора соли Мора с дихроматом калия. Ход определения а) Определение расхода КMпО4 на титрование соли Мора В колбу для титрования вносят пипеткой 3 мл раствора соли Мора и титруют из бюретки рабочим раствором КМпО4 до появления розовой окраски раствора. Титруют до получения трех сходящихся результатов.
Результаты измерений заносятся в таблицу 1: Таблица 1
Затем в колбу для титрования вносят пипеткой 2 мл раствора дихромата калия и 3 мл раствора соли Мора. Смесь перемешивают и оттитровывают избыток соли Мора раствором КMnO4 до появления серо-сиреневой окраски раствора от очередной капли KMnO4. Результаты измерений записываются в таблицу 2:
Таблица 2
На титрование остатка соли Мора, который не прореагировал с дихроматом калия пошло раствора перманганата калия:V’’(KMnO4) – V’(KMnO4). Тогда на основании закона эквивалентов можно записать: [V’’(KMnO4) – V’(KMnO4)] · C(1/5 KMnO4) = V(K2Cr2O7) · C(1/6 K2Cr2O7) Отсюда рассчитывается C(1/6 K2Cr2O7) и массу дихромата калия содержащегося в колбе. МЕТОД ЙОДОМЕТРИИ I. Сущность метода В этом методе используются окислительные свойства свободного йода и восстановительные свойства йодид-ионов: I2 + 2e = 2I- φo = + 0,54 B 1. Применяя рабочий раствор йода, можно определить количество различных восстановителей, окислительно-восстановительный потенциал которых меньше потенциала системы I2/2I-. 2. Используя растворы йодидов, например KI, можно определять количество окислителей, окислительный потенциал которых выше, чем потенциал системы I2/2I-. Окисление восстановителей производят непосредственным титрованием раствора восстановителя рабочим раствором йода. Примером может служить определение сульфита натрия, который реагирует с I2 по уравнению: I2 + Na2SO3 + H2O = Na2SO4 + 2HI Аналогичным образом можно определить количество SnCl2, H2S и сульфидов, H3AsO3. и других восстановителей. Однако йодометрическсе определение восстановителей прямым титрованием рабочим раствором иода не находит широкого применения. Гораздо чаще их определяют титрованием п о остатку. Для этого к раствору, содержащему восстановитель, прибавляют в избытке рабочий раствор I2, как это делают, например, при определении содержания серовсдорсда в сероводородной воде: H2S + 12 = S0 + 2HI Остаток I2 оттитровывают рабочим раствором тиосульфата натрия.Na2S2O3 по реакции: 2Na2S2O3 + I2 = Na2S4O6 + 2NaI f(Na2S2O3) = 1; f(I2) = 1/2. Наиболее широкое применение иодометрический метод находит для определения количества окислителей по методу замещения. Так определяют перманганат, дихромат, Cu2+, Fe3+, ClO- и др. Например, при определении количества КMпО4 к раствору перманганата в кислой среде приливают раствор КI (который является вспомогательным раствором), причем выделяется I2, количество которого эквивалентно содержанию КМпО4, (первая стадия): 2KMnO4 + 10KI + 8H2SO4 = 5I2 + 2MnSO4 + 6K2SO4 + 8H2O. f(KMnO4) = 1/5; f(KI) = 1. Выделившийся йод оттитровывают рабочим раствором тиосульфата натрия (вторая стадия): 2Na2S2O3 + I2 = 2NaI + Na2S4O6. Осуществить такое определение непосредственно титрованием окислителей раствором KI невозможно, т.к. не удается заметить окончание образования 12, резкого изменения внешнего вида раствора не наблюдается. Прямое титрование окислителей раствором тиосульфата натрия также невозможно, потому что реакция протекает очень сложно и образуется смесь продуктов неопределенного состава, вследствие чего нельзя произвести расчет. 3. Кроме окислителей и восстановителей методом йодометрии можно определять количество сильных кислот. Определениеих основано на том, что в нейтральной среде KI и КIO3 не взаимодействуют, но если к смеси KI + К103 добавить кислоты, то идет выделение свободного иода по уравнению: KIO3 + 5KI + 6HCl = 3I2 + 6KCl + 3H2O f(KIO3) = 1/5; f(KI) = 1; f(HCl) = 1/6. Из уравнения реакции следует, что кислота участвует в реакции и количество выделившегося йода эквивалентно количеству имевшейся в растворе кислоты. Определение точки эквивалентности Точку эквивалентности в йодометрии можно определить по появлению или исчезновению 12, водный раствор которого в присутствии КI довольно интенсивно окрашен в желто-коричневый цвет(комплекс KI3). Однако гораздо более точные результаты получается при введении индикатора - раствора крахмала. Крахмал с I3- адсорбционное соединение темно-синего цвета. В этом соединении молекула йода деформирована, благодаря чему она меняет свою окраску из коричневой в темно-синию. При непосредственном титровании восстановителей. например SnCI2, Na2HAsO3 и др., крахмал прибавляют перед началом титрования. При титровании восстановителей по остатку, а также при определении окислителей и кислот по методу замещения крахмал прибавляют к реакционной смеси в конце титрования, когда раствор из бурого станет бледно-желтым. Иначе крахмал будет адсорбировать большие количества йода и медленно его отдавать в раствор, вследствие чего будет затрачен избыток тиосульфата натрия и искажены результаты титрования. Условия выполнения иодометрических определений 1) Свободный йод летуч и при нагревании летучесть его увеличивается. Чувствительность крахмала как индикатора (адсорбция) с повышением температуры понижается. Поэтому титрование следует вести на холоду. 2) Иод реагирует со щелочами согласно уравнению: 3I2 + 6NaOH = NaIO3 + 5NaI + 3H2O Поэтому иодометрические определения нельзя проводить в сильно- щелочной среде. Результат анализа при этом не может быть точным. 3) Реакции, протекающие при иодометрических определениях, являются не очень быстрыми. Для увеличения скорости реакции иодида калия с окислителем берется избыток серной кислоты и иодида. Реакционной смеси дают постоять 5-6 мин 4) Конец титрования устанавливают по исчезновению (или появлению) синей окраски, присущей крахмалу в присутствии свободного йода. Следует помнить, что раствор должен обесцветиться от одной капли тиосульфата натрия. Дальнейшее прибавление последнего не изменит окраски титруемого раствора, но сделает анализ неверным. С помощью йодометрических титровании находят количество сахара в крови, определяют константу скорости реакции окисления KI действием Н2О2 и т.д. Приготовление рабочих растворов В иодометрии используется два рабочих раствора: I2 и Na2S2O3, а также вспомогательный раствор KI(произвольной концентрации – обычно 10%). а) Рабочий раствор йода. Свободный йод, очищенный возгонкой, удовлетворяет требованиям, предъявляемым к исходным веществам, поэтому титрованный раствор его готовят растворением точной навески I2 в водном растворе KI. При этом происходит образование комплексного соединения: KI + I2 = K[I3] Образование комплекса не отражается на скорости взаимодействия рабочего раствора иода с восстановителем из-за малой прочности [I3]- б) Рабочий раствор тиосульфата натрия В отличие от йода тиосульфат натрия Na2S2O3 · 5H2O является сравнительно нестойким веществом, и при растворении его в дистиллированной воде, в которой имеется углекислый газ, происходит следующая реакция: Na2S2O3 + CO2 + H2O = NaHCO3 + NaHSO3 + S Этот процесс идет очень медленно, т.к. концентрация СО2 невелика (закон действия масс), и заканчивается дней через 10. Тиосульфат натрия может также разлагаться тиобактериями и окисляться кислородом воздуха. Поэтому необходимо: а) взвешивать рассчитанное количество Na2S2O3 · 5H2O на технохимических весах; б) растворять в свежепрокипяченной и охлажденной без доступа CO2 дистиллированной воде; в) определять концентрацию раствора не раньше, чемчерез 10 дней; г) защищать раствор тиосульфата от углекислого газа и света(способствует размножению тиобактерий). В качестве исходного вещества для установления концентрации раствора Na2S2O3 обычно применяют дихромат калия K2Cr2O7. Лабораторная работа ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ РАСТВОРА Na2S2O3 по K2Cr2O7 Kак уже отмечалось выше, непосредственное титрование раствора дихромата раствором тиосульфата натрия невозможно, т.к образуется несколько продуктов реакция – Na2SO4,Na2S4O6 и другие, т.е. не соблюдается одно из основных требований к реакциям объемного анализа. Титрование ведут по методу замещения. Реакция идет в две стадии: I) Дихромат калия – К2Сг2О7,, взаимодействуя с KI, выделяет эквивалентное количество I2 в соответствии с уравнением: K2Cr2O7 + 6KI + 7H2SO4 = Cr2(SO4)3 +4K2SO4 + 3I2 + 7H2O f(K2Cr2O7) = 1/6; f(KI) = 1. 2) Выделившийся I2 оттитровывают раствором Na2S2O3 – количество Na2S2O3, пошедшее на титрование, эквивалентно вступившему в реакцию количеству K2Cr2O7. I2 + Na2S2O3 = 2NaI + Na2S4O6 f(I2) = 1/2; f(Na2S2O3) = 1. В соответствии с законом эквивалентов: C(1/6 K2Cr2O7) · V(K2Cr2O7) = C(Na2S2O3) · V(Na2S2O3) Приготовление раствора исходного вещества - дихромата калия по точной навеске Расчет навески m(K2Cr2O7) = C(1/6 K2Cr2O7) · M(1/6 K2Cr2O7) · Vколбы Например, необходимо приготовить 250 мл 0,02н K2Cr2O7. m(K2Cr2O7) = 0,02 · 49,04 · 0,25 = 0,2452 г Порядок титрования В бюретку наливают раствор тиосульфата натрия. В колбу для титрования – растворы дихромата калия, серной кислоты и иодида калия. Индикатор - раствор крахмала (добавляет в конце титрования). Ход определения В колбу для титрования пипеткой переносят из мерной колбы 3 мл раствора K2Cr2O7, прибавляют 3мл раствора KI и 3 мл 2н раствора Н2SO4, отмеривая их мерной пробиркой или пипеткой. Колбу закрывают часовым стеклом и дают стоять 5 минут, защищая от прямых солнечных лучей. По истечении указанного срока (не ранее) к реакционной смеси добавляют 10 мл дистиллированной воды и титруют раствором тиосульфата натрия до светло-желтой окраски. Затем прибавляют 0,5 мл раствора крахмала (10 капель). Раствор при этом окрасится в интенсивно синий цвет. Не фиксируя расход Na2S2O3 и не доливая раствор его в бюретку, продолжают титрование до исчезновения синей окраски. Во время титрования необходимо энергично размешивать содержимое колбы, чтобы уловить тот момент, когда от одной капли раствора Na2S2O3 синяя окраска исчезнет. Нужно помнить, что раствор при этом не станет совсем бесцветным, а будет слабо окрашен в светло-зеленый цвет, присущий ионам Cr3+(точнее [Cr(H2O)6]3+ ), возникающим в результате реакции Титрование повторяют до получения сходящихся результатов и производят расчет концентрации тиосульфата натрия. Результаты экспериментов оформляются в виде таблицы:
С(Na2S2O3) = C(1/6 K2Cr2O7) V(K2Cr2O7) / V(Na2S2O3) Лабораторная работа ОПРЕДЕЛЕНИЕ МАССЫ ХЛОРАМИНА Б Хлорамин Б – кристаллогидрат N-хлорбензолсульфамида натрия C6H5SO2N(Cl)Na · 3H2O. M (C6H5SO2N(Cl)Na · 3H2O) = 267,5 г/моль.
2% раствор хлорамина Б в воде используется в целях дезинфекции. При растворении хлорамина Б в воде образуется гипохлорит-ион ClO-: C6H5SO2N(Cl)Na + H2O = C6H5SO2NH2 + ClO- + Na+ Дезинфицирующее действие гипохлорит-иона объясняется его сильной окислительной способностью: ClO- + H2O + 2e = Cl- + 2OH- φo = + 0,88 B Определение гипохлорит-иона, эквивалентное содержанию хлорамина Б, также как и других окислителей, основано на следующих реакциях: ClO- + 2I- + 2H+ = I2 + Cl- + H2O f(хлорамин Б) = 1/2 I2 + S2O32- = 2I- + S4O62- Ход определения Раствор хлорамина Б получают от преподавателя в мерной колбе на 100 мл, содержимое колбы разбавляют дистиллированной водой до метки и тщательно перемешивают. В колбу для титрования пипеткой переносят из мерной колбы 3 мл раствора хлорамина Б, прибавляют 3мл раствора KI и 3 мл 2н раствора Н2SO4, отмеривая их мерной пробиркой или пипеткой. Колбу закрывают часовым стеклом и дают стоять 5 минут, защищая от прямых солнечных лучей. По истечении указанного срока (не ранее) к реакционной смеси добавляют 10 мл дистиллированной воды и титруют раствором тиосульфата натрия до светло-желтой окраски. Затем прибавляют 0,5 мл раствора крахмала (10 капель). Раствор при этом окрасится в интенсивно синий цвет. Не фиксируя расход Na2S2O3 и не доливая раствор его в бюретку, продолжают титрование до исчезновения синей окраски. Во время титрования необходимо энергично размешивать содержимое колбы, чтобы уловить тот момент, когда от одной капли раствора Na2S2O3 синяя окраска исчезнет. Титрование повторяют до получения сходящихся результатов. Результаты опытов представляются в виде таблицы:
C(1/2 C6H5SO2N(Cl)Na ∙ 3H2O)∙ V(C6H5SO2N(Cl)Na ∙ 3H2O) = C(Na2S2O3) ∙Vср.(Na2S2O3) Отсюда C(1/2 C6H5SO2N(Cl)Na ∙ 3H2O) = m(хлорамин Б) = C(1/2 C6H5SO2N(Cl)Na ∙3H2O) ∙ M(1/2 C6H5SO2N(Cl)Na ∙3H2O) ∙ V(колбы).
ХИМИЧЕСКИЕ МЕТОДЫ
Эти методы основаны на выполнении некоторых химических реакций между исследуемым веществом и реактивами, в результате чего можно произвести измерение количества определяемого вещества. Основными химическими методами являются весовой (гравиметрия) и объемный (титриметрия). Весовой метод Определяемый элемент в навеске анализируемого вещества при помощи химической реакции переводят в осадок. Полученный осадок отфильтровывают, промывают, высушивают (если необходимо, прокаливают) и взвешивают. Зная массу исходного вещества, массу и состав полученного осадка, можно рассчитать содержание определяемого элемента в исследуе
|
||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 419; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.118.24.176 (0.01 с.) |