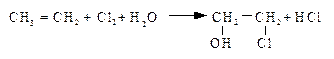Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Гидроформилирование олефинов.Содержание книги
Поиск на нашем сайте Реакция оксосинтеза дает возможность в зависимости от исходного олефина получать разнообразные альдегиды. Из этилена получается только один альдегид— пропионовый: CH2 = CH2 + CO +H2 → CH3CH2CHO В качестве побочного продукта, особенно при более низких температурах (например, при 50°С), образуется диэтилкетон: 2СН2=СН2 + СО + Н2 → С2Н5СОС2Н5 Остальные олефины дают альдегиды нормального и изостроения, например из пропилена образуются н-масляный и изомасляный альдегиды:
CH3CHCH3 CHO Соотношение альдегидов нормального и изостроения колеблется от 1,5: 1 до 3,5: 1. Можно повысить это соотношение путем изменения условий реакции и модификации катализаторов: факторы, замедляющие реакцию (повышение парциального давления СО и понижение температуры), способствуют росту этого соотношения, а факторы, ускоряющие реакцию (повышение температуры и парциального давления водорода), способствуют его снижению. Катализаторы гидроформилирования. Гидроформилирование представляет собой гомогеннокаталитический процесс, протекающий в присутствии комплексных соединений кобальта, в частности дикобальтоктокарбонила Со2(СО)8 и гидротетракарбонила кобальта НСо(СО)4 (привычные названия — карбонил кобальта, гидрокарбонил кобальта), образующихся по реакциям:
Характерной особенностью этих комплексных соединений является их нестабильность: в отсутствие оксида углерода карбонил кобальта разлагается при температурах выше 50°С, а гидрокарбонил кобальта—при температурах ниже 0°С:
Их стабильность при высоких температурах процесса может быть обеспечена только при высоком парциальном давлении оксида углерода, так как с повышением давления равновесие этих реакций смещается влево. В качестве катализаторов гидроформилирования предложены также родиевые катализаторы. Активность родия, его карбонилов, фосфиновых и арсиновых комплексов в 100-1000 раз более высокая, чем карбонилов кобальта, она проявляется при 75-150оС и давлении 5-20 МПа. Однако они недостаточно стабильны и разрушаются в ходе реакции. Стабильность и активность карбонилов родия как катализаторов гидроформилирования повышаются при их модификации фосфинами, фосфитами, арсинами или аминами. Влияние основных факторов на процесс гидроформилирования. Соотношения Н2:СО. Увеличение соотношения Н2:СО повышает скорость реакции, его обычно варьируют в пределах 1: 1—2: 1. Температура. Скорость гидроформилирования растет с повышением температуры. Повышение температуры влияет не только на скорость, но и на состав продуктов: растет выход альдегидов изостроения и ускоряется гидрирование альдегидов в спирты. Реакция подчиняется уравнению Аррениуса в интервале 120—180 °С. Отклонение скорости реакции от уравнения Аррениуса при температурах выше 180 °С объясняется тем, что при этих условиях начинается разложение карбонилов кобальта. Давление. Общее давление в системе зависит от парциальных давлений СО и Н2. Увеличение парциального давления СО сверх определенного предела тормозит реакцию гидроформилирования, а повышение парциального давления Н2 ускоряет ее. Общее давление в системе составляет обычно 10—30 МПа. Необходимо отметить, что каждой температуре отвечает определенное давление, выше которого скорость процесса перестает от него зависеть. Такую закономерность можно объяснить сложным характером влияния парциальных давлений СО и Н2.
ВАРИАНТЫ ТЕХНОЛОГИЧЕСКОГО ОФОРМЛЕНИЯ СТАДИИ ГИДРОФОРМИЛИРОВАНИЯ К сырью для оксосинтеза предъявляются жесткие требования в отношении содержания примесей. Из-за того, что диеновые и ацетиленовые углеводороды образуют с карбонилами кобальта неактивные комплексы, появляется индукционный период гидроформилирования, когда катализатор регенерируется, а диеновый или ацетиленовый углеводород гидрируется до олефина. Наличие в синтез-газе кислорода приводит к разложению карбонила кобальта с образованием неактивного оксида СоО, что также замедляет реакцию. Пероксидные соединения также взаимодействуют с гидрокарбонилами, замедляя гидроформилирование и обусловливая большой индукционный период. Поэтому сырье нужно подвергать предварительной очистке. Процесс гидроформилирования является экзотермическим: тепловой эффект равен 117 кДж/моль и мало зависит от молекулярной массы и строения углеводорода. Для процесса имеет большое значение эффективный теплоотвод и поддержание стабильного температурного режима. Отвод тепла осуществляется несколькими способами: 1) в реакторе монтируют трубчатый холодильник, в межтрубном пространстве которого циркулирует вода или синтез-газ—он при этом нагревается до нужной температуры; 2) наряду с внутренним охлаждением применяются также выносные холодильники; 3) в реактор возвращают охлажденные продукты: за счет их нагревания отводится выделяющееся тепло. Реакция протекает при 120—170 °С и 15—30 МПа. Объемную скорость подачи олефина можно менять от 0,4 до 2 ч-1, что соответствует среднему времени контакта 1 ч. В промышленных условиях соотношение СО: Н2 поддерживается в интервале от 1:1 до 2:1, а концентрация катализатора равна 0,02—0,2% (масс.) в расчете на Со. Степень конверсии олефина зависит от его молекулярной массы и колеблется в пределах 65—80 %. С повышением степени конверсии олефина возрастает роль вторичных реакций. Конверсию регулируют изменением объемной скорости. Выход целевых альдегидов составляет не менее 75—85 % от стехиометрического и зависит от молекулярной массы олефина. Процесс гидроформилирования ведут с рециркуляцией синтез-газа. В связи с рециркуляцией необходимо выводить из системы инертные примеси путем промывки синтез-газа циркулирующими продуктами реакции под давлением с последующей десорбцией абсорбированных примесей и дополнительной отдувкой части циркулирующего газа. Гидроформилирование низших олефинов ведут в растворе углеводородов, обычно в растворе высококипящей фракции, остающейся после гидриро- вания и отделения спиртов. Продукты реакции потом легко отделяют от растворителя. Оксид углерода и водород барботируют через слой жидкости, что обеспечивает хорошее перемешивание. Специфической особенностью процесса является необходимость декобальтизации—извлечения карбонилов кобальта из реакционной смеси и возвращения их в процесс. Это основная технологическая трудность процесса, так как декобальтизация технологически значительно сложнее собственно гидроформилирования, и она определяет технико-экономические показатели процесса в целом. По способу декобальтизации технологические схемы оксосинтеза можно классифицировать следующим образом: 1) схемы с термическим разложением карбонилов кобальта, основанные на термической нестабильности карбонилов; 2) солевые схемы, основанные на нестабильности карбонилов кобальта к действию окислителей; 3) испарительные схемы, основанные на различной летучести карбонилов кобальта и продуктов оксосинтеза. 4) смешанные схемы, сочетающие принципы солевых и испарительных схем. Схемы с термическим разложением карбонилов кобальта. Эти схемы основаны на обратимости реакции:
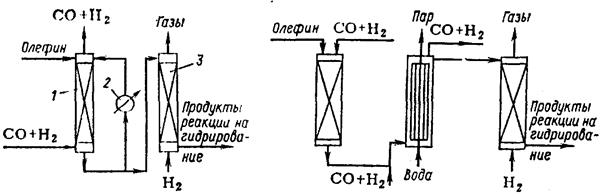
Как указывалось выше, образование карбонилов кобальта идет при более низкой температуре и достаточно высоком парциальном давлении СО, а распад их протекает при более высокой температуре и низком парциальном давлении СО. Как видно из уравнения, при разложении карбонилов образуется металлический кобальт, который обычно отлагается на твердых поверхностях. Это недопустимо, так как кобальт накапливается на стенках аппаратуры и не может быть возвращен в цикл. Поэтому в схемах с термическим разложением карбонилов кобальта в реактор вводят твердый носитель (кизельгур или пемзу), чтобы кобальт оставался на его поверхности. Существуют три разновидности этого способа: 1) с суспендированным носителем (кизельгурная схема); 2) со стационарным слоем носителя; 3) без носителя, с тонкодисперсным металлическим кобальтом. Схема с суспендированным слоем носителя (кизельгурная схема). Катализатор вводят в виде суспензии мелкозернистого кизельгура, на котором осажден металлический кобальт; суспензию готовят в специальном аппарате. Процесс гидроформилирования осуществляют в двух последовательно расположенных реакторах 1 и 2. Суспензию катализатора подают в реактор 1 одновременно с олефином и синтез-газом. Реактор работает под давлением 25—30 МПа при температуре 150— 160°С.
Кизельгурная схема оксосинтеза: 1,2 — реакторы карбонилообразования и гидроформилирования; 5 — реактор гидроформилирования; 3, 4—газосепараторы; 5 — реактор декобальтизации; 6—магнитный сепаратор. В нем образуются карбонилы кобальта и протекает реакция гидроформилирования до степени превращения олефина около 70%. В реакторе 2 реакция гидроформилирования завершается при 160—170 °С с глубиной 80% и более. Продукты реакции поступают в газосепараторы 3 и 4, где суспензия отделяется от газа и направляется в реактор декобальтизации 5, работающий при давлении водорода 2,5—3 МПа и температуре 120—130°С. В этих условиях карбонилы кобальта разлагаются и осаждаются на кизельгур. Жидкие продукты реакции отделяются от катализатора в магнитных сепараторах 6, после чего они направляются на гидрирование, а катализатор вновь поступает в реактор гидроформилирования. Такая схема осуществлена в промышленном масштабе. Характерной особенностью приведенной схемы является использование для гидрирования альдегидов в спирты кобальта, осажденного на мелкозернистом кизельгуре. Схемы со стационарным слоем носителя. В промышленности применяются две разновидности схем—двухреакторную и триадную. В колонне оксосинтеза 1 помещен стационарный слой пемзы, на которой осажден металлический кобальт. Жидкий олефин подают в колонну сверху, а синтез-газ снизу, и они движутся противотоком. В колонне 1 непрерывно происходит образование карбонилов кобальта и растворение их в олефине. В результате реакции выделяется тепло, которое отводится в выносном холодильнике 2, а охлажденные продукты возвращаются в колонну. Часть продуктов поступает в декатализер 3, куда подается водород и где благодаря низкому парциальному давлению СО карбонилы кобальта разлагаются. Выделяющийся при этом металлический кобальт осаждается на насадке, которой заполнен аппарат. После истощения катализатора в колонне 1 и накопления кобальта в декатализере 3 направление потоков меняется на обратное.
Схема двухреакторного реакционного узла оксосинтеза: 1 — колонна оксосинтеза; 2 — холодильник; 3 — декатализер. Реакторный блок триадной схемы оксосинтеза: 1 — катализер; 2—колонна оксосинтеза; 3 — декатализер Триадная схема отличается тем, что реакторный блок состоит из трех аппаратов. Металлический кобальт осажден на стационарном носителе в катализере 1. Жидкий олефин и синтез-газ подаются сверху; при этом образуются карбонилы кобальта, которые растворяются в олефине. Температура в катализере поддерживается 150—180°С, давление 15—30 МПа. Раствор катализатора в олефине вместе с синтез-газом поступает в реактор 2. Туда же дополнительно подают синтез-газ. Отвод выделяющегося тепла осуществляется при помощи вмонтированного в аппарат трубчатого холодильника, в котором генерируется водяной пар. Декобальтизация жидких продуктов осуществляется в декатализере 3, который устроен так же, как катализер 1. В декатализер подают водород. При 150—180°С и низком парциальном давлении СО происходит разложение карбонилов кобальта, и металлический кобальт осаждается на насадке. После истощения кобальта в катализере и накопления его в декатализере направление потоков меняется на обратное. В порошковой схеме используется металлический кобальт, который вводят в реактор в виде тонкого порошка. Карбонилы кобальта разлагаются в декатализере, куда подают рециркулят с суспендированным порошком металлического кобальта; на этот порошок и высаживается продукт разложения. Суспензию после отделения продуктов реакции возвращают в реактор оксосинтеза. Основные недостатки схем с термическим разложением карбонилов кобальта следующие: - большой удельный расход металла, так как разложение карбонилов кобальта осуществляется при высоком давлении; - эрозия аппаратуры и измельчение носителя в схемах с суспендированным катализатором; - периодичность работы реакторов в схеме со стационарным слоем катализатора, что требует сложной автоматики; - отложение кобальта на поверхности аппаратуры, особенно в схемах с суспендированным катализатором. Солевые схемы основаны на разложении карбонилов кобальта окислителями. Так, кобальт вводят в реактор в виде солей жирных или нафтеновых кислот, из которых в условиях гидроформилирования образуются карбонилы:
Распад и образование карбонилов кобальта представляют собой окислительно-восстановительную реакцию. Реакцию разложения карбонилов ведут в присутствии окислителей; без них она идет слишком медленно. Температура на стадии разложения не должна быть высокой во избежание разложения карбонилов до металлического Со. Для процесса выбирают такие кислоты, которые образуют водорастворимые соли, легко отделяемые от декобальтизованного продукта (минеральные или низшие карбоновые кислоты). Водорастворимую соль кобальта затем переводят в маслорастворимую, например нафтенат кобальта, и в таком виде подают в систему образования карбонилов. В процессе образования карбонилов выделяются соответствующие карбоновые кислоты. Более перспективными оказались смешанные схемы, в которых сочетается принцип солевых и испарительных схем. От чисто солевых схем они отличаются тем, что карбонилы кобальта переводят в маслорастворимую соль, например нафтеновой кислоты, являющуюся термоустойчивой и малолетучей. От раствора кобальтовой соли отгоняют продукты гидроформилирования, а остаток возвращают на стадию кобальтизации, где опять образуются карбонилы кобальта.
ПОЛУЧЕНИЕ ОКСИДОВ ОЛЕФИНОВ Промышленное производство оксидов олефинов ограничивается этилен- и пропиленоксидами. Оксид этилена является многотоннажным промышленным продуктом. На основе оксида этилена получают: этиленгликоль, используемый как антифриз, этаноламины, применяемые в качестве абсорбентов для удаления кислых примесей из газов, неионогенные синтетические моющие вещества. Известны два основных способа производства оксида этилена: классический хлоргидринный и современный метод прямого окисления этилена. Получение оксида этилена хлоргидридным методом состоит из двух стадий. На первой стадии при пропускании этилена и хлора через воду образуется этиленхлоргидрин:
Температура реакции 48—52°С, давление около 1,1 атм. На второй стадии полученный раствор этиленхлоргидрина омыляется раствором известкового молока (10—12%-ной концентрации) при температуре 90—100°С, который приливают к этиленхлоргидрину:
Оксид этилена отгоняется затем из водного раствора. Экономически более выгодным является получение оксида этилена прямым окислением этилена, так как при этом методе не тратится хлор. Себестоимость оксида этилена, полученного прямым окислением, на 20% ниже, чем при получении его хлоргидринным методом. Прямое окисление этилена протекает над серебряным катализатором при температуре 220—280°С и давлении 7—10 атм. Реакция протекает в критических условиях; если условия мягкие, образуется мало оксида этилена, в жестких условиях этилен сгорает с образованием диоксида углерода и воды. В качестве окислителя используют кислород воздуха или чистый кислород. Окисление этилена протекает по реакции:
Побочная реакция — глубокое окисление этилена до диоксида углерода с выделением большого количества тепла:
Для окисления применяется 98%-ный этилен, который смешивается с воздухом; концентрация этилена в реакционной смеси составляет при этом 3%. Время контакта с катализатором 1,5 с. Процесс окисления ведется, как правило, на стационарном катализаторе, реже — в кипящем слое. Пропиленоксид также является крупнотоннажным продуктом, основное количество которого идет на получение пропиленгликоля и полимерных материалов – полипропиленоксидов, пенополиуретанов и др. На его основе получают также деэмульгаторы (проксанолы, проксамины) для обезвоживания и обессоливания нефти, а также неионогенные моющие средства. Из него легко можно получить глицерин через стадию образования аллилового спирта, который, в свою очередь, может быть использован для получения ненасыщенных полиэфиров. Основными промышленными процессами получения пропиленоксида являются хлоргидринный метод и окисление пропилена пероксидами углеводородов. Первый процесс аналогичен рассмотренному для производства этиленоксида – получающаяся на первой стадии смесь пропиленхлоргидринов при омылении дает пропиленоксид. Гипохлорирование проводят при температуре 35-45оС и атмосферном давлении, выход пропиленхлоргидринов составляет 87-90%, а основного побочного продукта дихлорпропана 6-9%. Для омыления пропиленхлоргидринов используется известковое молоко или 1-%-ный раствор хлорной извести. Пропиленоксид выделяют ректификацией, выход его составляет 95% на пропиленхлоргидрин. Основными недостатками этого процесса являются: большие количества отходов в виде шлама, значительные расходы хлора, щелочи, хлорной извести и пара. Процессы прямого окисления пропилена дают невысокие выходы пропиленоксида, поэтому в отличие от окисления этилена не получили промышленного развития. Промышленную реализацию получил процесс окисления пропилена гидропероксидом этилбензола, позволяющий получить наряду с пропиленоксидом еще один ценный продукт – стирол. Реакция эпоксидирования протекает с большой скоростью и селективностью в присутствии растворимых катализаторов – солей молибдена, вольфрама, ванадия, титана и др. Наиболее эффективны нафтенаты молибдена и вольфрама. Процесс проводят в жидкой фазе, при 2-5-кратном избытке олефина по отношению к гидропероксиду, при температуре 80-110оС. В зависимости от температуры, концентрации катализатора и природы исходных реагентов время реакции изменяется от 0,3 до 2 ч. Процесс состоит из трех стадий: ¯ Получение гидропероксида этилбензола окислением этилбензола:
На этой стадии процесса побочными продуктами являются ацетофенон и метилфенилкарбинол, образующиеся вследствие разложения гидропероксида этилбензола:
¯ Эпоксидирование пропилена гидропероксидом этилбензола с получением пропиленоксида и метилфенилкарбинола
¯ Получение стирола дегидратацией метилфенилкарбинола
|
||
|
|
Последнее изменение этой страницы: 2016-04-23; просмотров: 826; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.102 (0.008 с.) |

 СН3СН2СН2CHO
СН3СН2СН2CHO 2 СН3СН = CH2 + СО + Н2
2 СН3СН = CH2 + СО + Н2

 2Со+8СО Со2(СО)8, Со2(СО)8 + Н2 2НСо(СО)4
2Со+8СО Со2(СО)8, Со2(СО)8 + Н2 2НСо(СО)4
 Со2(СО)8 2Со+8СО
Со2(СО)8 2Со+8СО
 2НСо(СО)4 2СО+8СО+Н2
2НСо(СО)4 2СО+8СО+Н2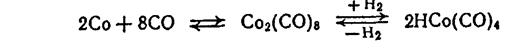

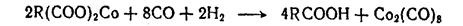
 Карбонилы после реакции разлагают серной кислотой в присутствии пероксида водорода:
Карбонилы после реакции разлагают серной кислотой в присутствии пероксида водорода: