Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Экспериментальные Методы исследованияСтр 1 из 15Следующая ⇒
Экспериментальные Методы исследования Адсорбции Лабораторные работы Утверждено Редакционным советом университета в качестве учебного пособия
Москва 2012 УДК 66.081.3(076) ББК 24.5я7 Э41
Авторы: М. Б. Алёхина, Т. В. Конькова, Е. Ю. Либерман, А. Г. Кошкин
Рецензенты: Доктор технических наук, профессор кафедры промышленной экологии и технологии неорганических веществ Московского Государственного Открытого Университета им. В. С. Черномырдина И.В. Семёнова Кандидат технических наук, зав. лабораторией кафедры технологии защиты биосферы РХТУ им. Д. И. Менделеева И.Н. Каменчук
Экспериментальные методы исследования адсорбции. Лабораторные работы: Э41 учеб. пособие /М. Б. Алёхина, Т. В. Конькова, Е. Ю. Либерман, А. Г. Кошкин − М.: РХТУ им. Д. И. Менделеева, 2012. − 88 с.
Учебное пособие представляет сборник лабораторных работ по адсорбционным методам, применяющимся в технологии неорганических веществ. Включает краткое изложение теоретических основ и описание ряда методов исследования адсорбентов и катализаторов. Предназначено для студентов специальности 240301.65 – Химическая технология неорганических веществ.
УДК 66.81.3(076) ББК 24.5я7 ISBN 978-5-7237-1062-7 © Российский химико-технологический университет им. Д. И. Менделеева, 2012 © Алёхина М. Б., Конькова Т. В., Либерман Е. Ю., Кошкин А. Г., 2012 Оглавление
Введение…………………………………………………………..………………5 1. Адсорбционное равновесие……………………………………………6 1.1. Основные положения теории адсорбции индивидуальных веществ………6 1.2. Основные уравнения, описывающие адсорбционное равновесие…….…..11 2. Экспериментальные методы измерения изотерм адсорбции……………………………………………………………….………20 2.1. Статические методы измерения изотерм адсорбции……………….……...21
2.1.1. Волюмометрический (объёмный) метод……………………….....…..21 2.1.2. Гравиметрический (весовой) метод………………………………...…23 2.1.3. Эксикаторный метод………………………………………………...…24 2.1.4. Метод переменных концентраций и переменных объёмов……….…25 2.2. Методы определения удельной поверхности твёрдых тел…………....…...25 2.2.1. Определение удельной поверхности по адсорбции красителей….....27 2.2.2. Определение удельной поверхности по изотермам адсорбции стандартного пара……………………………………………………………......…28 2.3. Лабораторные работы……………………………………………….....….…30 Лабораторная работа 1. Определение текстурных характеристик адсорбентов и катализаторов по изотерме адсорбции азота…………….......…..30 Лабораторная работа 2. Определение удельной поверхности твёрдых веществ методом тепловой десорбции азота…………………..……….35 Лабораторная работа 3. Определение изотермы адсорбции толуола на активном угле при 30 оС при помощи ТОЗМ и Банка данных по адсорбции………………………………………………………………………..40 Лабораторная работа 4. Измерение изотерм адсорбции паров воды на образцах адсорбентов и катализаторов эксикаторным методом…………….46 Лабораторная работа 5.Измерение изотерм адсорбции азота и кислорода при повышенном давлении……………………………...…………………….....…..…49 Лабораторная работа 6. Измерение изотермы адсорбции органического красителя из водных растворов на активном угле методом переменных концентраций……………………………………………………………………….54 3. Кинетика адсорбции……………………………………………....…......59 3.1. Основные положения теории кинетики адсорбции…………………..........59 3.2. Адсорбционно-кинетические методы изучения механизмов диффузии…60 3.3. Изучение самодиффузии адсорбированных молекул методом ядерного магнитного резонанса с импульсным грандиентом магнитного поля …………65 Лабораторная работа 7. Определение кинетических кривых адсорбции азота и кислорода при атмосферном давлении волюмометрическим методом………...67 4. ТЕРМИЧЕСКИЙ АНАЛИЗ СИСТЕМ……………………………………....….72 4.1. Теоретическая часть….……………………………….………………….…..72
4.2. Дериватография……………………………………………..………..………78 4.3. Расчёт кинетических параметров процессов термического разложения....80 Лабораторная работа 8. Определение температурного режима первичной подготовки адсорбента (катализатора) и его термостойкости………..................82 ЛИТЕРАТУРА…………………………………………………………….….…….83 ПРИЛОЖЕНИЕ………………………………………………….……………........85 Введение Адсорбционные процессы широко используются в промышленности. Их применяют для очистки сырья, выделения целевых продуктов и их тонкой очистки, а также для обработки промышленных выбросов и выделения из них ценных компонентов. В настоящее время существует большое количество методов исследования состава и структуры поверхности адсорбентов: хроматографические, термодесорбционные и кинетические методы, изотопный обмен, инфракрасная спектроскопия, ЯМР, ЭПР, рентгеновские дифракционные методы, Оже-спектроскопия, электронная микроскопия высокого разрешения и т.д. Однако традиционные методы исследования адсорбции, несмотря на продолжительный опыт использования, остаются основными методами при изучении величин адсорбции разных компонентов, определения удельной поверхности и пористости адсорбентов. В пособии изложены методики проведения экспериментов, связанных с изучением адсорбционного равновесия и кинетики адсорбции, определением текстурных характеристик пористых тел на основе изотерм адсорбции. По представленным методикам предложено 8 лабораторных работ.
Адсорбционное равновесие Вопросы для самоконтроля 1. Какие существуют типы изотерм адсорбции? 2. Какое вещество является стандартным паром для определения текстурных характеристик пористых материалов? 3. Каково основное допущение при расчёте общего адсорбционного объема пор? 4. Почему нельзя определить объём макропор по изотерме адсорбции азота? 5. В чем сущность t -метода для определения поверхности супермикропор?
Лабораторная работа 2 Вопросы для самоконтроля 1. Какие существуют методы определения удельной поверхности веществ? Перечислите их достоинства и недостатки. 2. Каковы основные положения теории БЭТ? 3. В чём сущность метода тепловой десорбции азота? 4. Какие газы применяют для измерения удельной поверхности веществ в качестве адсорбтивов? Каковы требования к этим газам?
Лабораторная работа 3 Определение изотермы адсорбции толуола на активном угле с использованием теории объёмного заполнения микропор (ТОЗМ) и Банка данных по адсорбции Приборы и реактивы: − автоматическая адсорбционная установка Nova 1200e; − баллоны с азотом и гелием; − адсорбент – активный уголь АР-А; − адсорбтив – азот; − сосуд Дьюара с жидким азотом. Цель работы. Измерение изотермы адсорбции азота на активном угле АР-А при 77 К, расчёт параметров уравнения Дубинина−Радушкевича (W o, E o) и изотермы адсорбции толуола на АР-А при 303 К, проверка по Банку данных. Сущность работы. Имеющееся в наличии исследователя оборудование не всегда позволяет получить необходимые экспериментальные данные. Но благодаря ТОЗМ, из изотермы адсорбции стандартного пара (азота) на исследуемом образце путём расчёта можно получить изотерму адсорбции практически любого другого адсорбтива при нужном значении температуры [4,5]. Полученные расчетные данные можно сопоставить с данными для материала схожей природы, имеющимися в Банке данных [16].
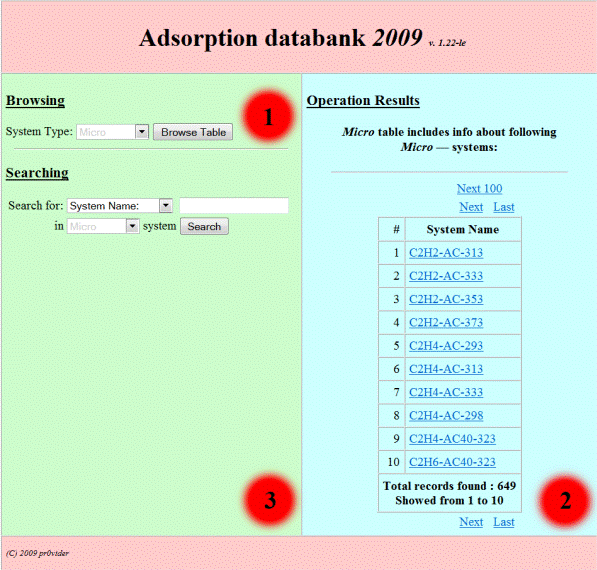
Банк данных по адсорбцииразработан в МГУ имени М. В. Ломоносова и является общедоступным через интернет банком данных в форме сайта http://www.adsorption.ru. Он представляет собой интерактивную систему просмотра и поиска адсорбционных данных. В настоящее время в Банке содержатся более 110 изотерм адсорбции индивидуальных паров и газов на макропористых адсорбентах, более 760 изотерм на микропористых адсорбентах, более 80 изотерм адсорбции компонентов растворов на макропористых адсорбентах, более 60 изотерм адсорбции компонентов растворов на микропористых адсорбентах, более 100 изотерм адсорбции смесей газов на различных адсорбентах, 10 тройных систем, данные более чем по 230 адсорбентам, производящимся в Российской Федерации и за рубежом. Банк постоянно пополняется. Для удобства пользователей в Банке приводятся результаты обработки экспериментальных изотерм адсорбции паров и компонентов бинарных растворов в соответствии с уравнениями, константы которых дают важную информацию о соответствующих адсорбционных системах. В Банке данных представлены константы уравнений, по которым проводилась обработка, табличные данные экспериментальных изотерм адсорбции и соответствующие экспериментальные и расчётные графики. Характеристики адсорбентов соответствуют опубликованным в специализированных каталогах или технологическим данным фирм-производителей. На рис. 2.9 и 2.10 приводятся формы просмотра в Банке данных.
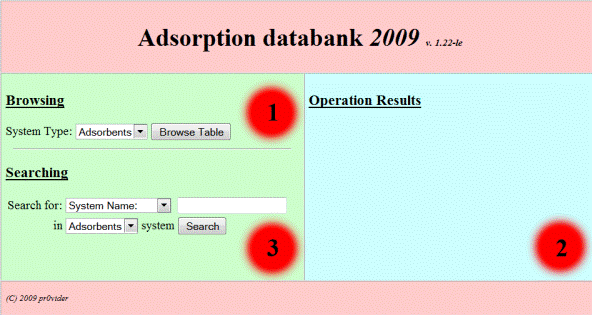
Рис. 2.9. Вид стартовой формы построения запросов в Банке данных
Основное окно (рис. 2.9) разделено на 3 зоны: 1. Зона выбора параметров просмотра в базе. 2. Зона вывода результатов запросов. 3. Зона выбора параметров поиска в базе. Для просмотра данных необходимо в первой зоне выбрать интересующий раздел базы (таблицу) из следующего списка: Adsorbents (данные об адсорбентах), Binvapor (экспериментальные данные изотерм адсорбции смесей газов на различных адсорбентах), Binsmi (экспериментальные данные изотерм адсорбции из растворов на микропористых адсорбентах), Binsma (экспериментальные данные изотерм адсорбции из растворов на макропористых адсорбентах), Macro (экспериментальные данные изотерм адсорбции индивидуальных газов/паров на макропористых адсорбентах),
Micro (экспериментальные данные изотерм адсорбции индивидуальных газов/паров на микропористых адсорбентах), Ternary (экспериментальные данные изотерм адсорбции тройных смесей растворов/газов на различных адсорбентах). Выбрав нужный раздел базы из указанного списка, нажать кнопку “Browse table”.
После этого во второй зоне будет выведена таблица, приведенная на рис. 2.10 (для примера выбрана таблица индивидуальной адсорбции соединений на микропористых адсорбентах − Micro). Во второй зоне исходной формы выведена таблица найденных систем. Пользователь может перемещаться по таблице с помощью ссылок управления таблицей (Next, Next100, Last, Previous, Previous100, First), либо выбрать интересующую систему, кликнув кнопкой мыши на соответствующей ссылке. В результате вся первичная информация о системе будет представлена в отдельном окне.
Рис. 2.10. Вид формы просмотра таблицы данных на примере адсорбции индивидуальных компонентов на микропористых адсорбентах (раздел Micro) в Банке данных
Порядок проведения работы аналогичен изложенному в лабораторной работе 1. Температура регенерации в случае микропористых углей составляет 350 оС. В ходе опыта получают 10-15 равновесных точек на всем интервале изменений Р/ Ps. С помощью программы, заложенной в компьютерном обеспечении установки, экспериментальные данные обрабатываются по уравнению Дубинина−Радушкевича и определяются текстурные константы образца W o и E o для стандартного пара (азота). После этого, используя справочные данные [17], с помощью компьютера проводят расчет изотермы адсорбции толуола (С6Н5СН3) на угле АР-А. Результаты экспериментов и расчётов оформляются в виде таблиц, образцы которых приведены ниже (табл. 2.2. и табл. 2.3). Таблица 2.2 Порядок проведения работы Бюксы высушивают до постоянной массы при температуре (100±5) °С. Высушивание бюкса до постоянной массы считают законченным, когда два последовательных взвешивания дают одинаковые результаты. За массу высушенного бюкса принимают наименьшую величину полученную при взвешивании. Каждый образец помещают в отдельный высушенный бюкс. Взвешивают бюкс с образцом с погрешностью 2∙10-4 г. Бюксы с образцами высушивают до постоянной массы при выбранной температуре дегидратации (±5 °С). Результаты взвешиваний фиксируются в табл. 2.4 с указанием номера образца и температуры дегидратации.
Таблица 2.4 Результаты измерений
Края 10 эксикаторов и их крышек смазывают вакуумной смазкой для предотвращения попадания наружного воздуха во внутрь эксикатора. Наливают в каждый из 10 эксикаторов насыщенные растворы солей или водные растворы серной кислоты. В каждый эксикатор с растворами помещают по 2 бюкса с двумя образцами материала. Бюксы ставят на фарфоровую вставку эксикатора открытыми. Их крышки помещают рядом с бюксами. Эксикатор закрывают крышкой. Все бюксы необходимо помещать в эксикатор в один день. Эксикаторы с образцами материала размещают на полках лабораторного термостата, в которых поддерживают температуру (20±2) °С.
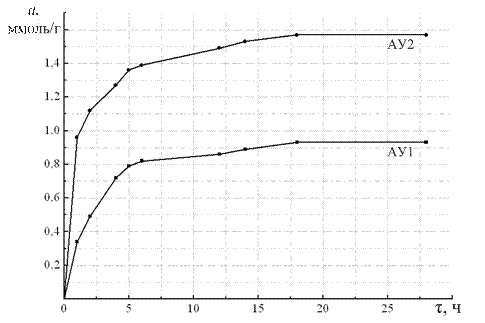
Бюксы с образцами материала взвешивают через каждый час, а затем – через каждые 12 ч до достижения образцами постоянной массы. Перед взвешиванием открывают эксикатор и сразу закрывают крышками все находящиеся в нём бюксы с образцами. Процесс поглощения материалом паров воды из окружающего воздуха (сорбции) считают законченным, когда два последовательных взвешивания дают одинаковые результаты или масса бюкса с образцом материала начнет уменьшаться. За массу бюкса с образцом материала после окончания процесса сорбции принимают наибольшую величину, полученную при взвешивании. По окончании процесса взвешивания для каждого значения относительной влажности получают кинетическую кривую адсорбции. Пример кинетической кривой приведен на рис. 2.11.
Рис. 2.11. Кинетические кривые адсорбции паров воды на образцах активированных углей
Равновесную величину адсорбции по парам воды образца материала, а Н2Ов г/100 г образца вычисляют по формуле: где m 1 – масса бюкса с образцом материала после окончания процесса сорбции, г; m 2– масса бюкса с образцом материала после высушивания образца до постоянной массы, г; m 3– масса высушенного до постоянной массы бюкса, г. По экспериментальным данным строят изотерму адсорбции паров воды при 20 оС. Пример изотермы адсорбции паров воды на образце активированного угля приведен на рис. 2.12.
Рис. 2.12. Изотермы адсорбции паров воды на образце АУ1, полученные эксикаторным методом и на вакуумно-сорбционной установке
После обработки результаты заносятся в табл. 2.5,по данным которой строится изотерма адсорбции паров воды на исследуемых образцах при Таблица 2.5 Вопросы для самоконтроля 1. В чём суть эксикаторного метода определения изотерм адсорбции? 2. Какие адсорбтивы можно использовать в этом методе? 3. Какова методика приготовления растворов насыщенных солей? Лабораторная работа 5 Объём адсорбента, л - Температура, °С - Барометрическое давление, мм рт.ст. - Газ – гелий Таблица 2.6 Результаты измерений
По результатам измерений строят калибровочную кривую зависимости свободного объёма системы от давления V св = f(P). Измерение изотермы адсорбции азота При закрытых кранах 2 и 10 и открытых кранах 3 и 11 с помощью вакуум-насоса 7 из адсорбера 1 откачивают воздух до остаточного давления Замерив объём десорбированного газа при давлении в адсорбере, равном 1 ата, подсоединяют газосчётчик 8 к выхлопу вакуум-насоса, открывают кран 3 при закрытых кранах 2 и 10 и откачивают газ из адсорбера при помощи вакуум-насоса 7, замеряя объём десорбата (кран 12 открыт). Все значения давления в адсорбере и соответствующие им показания газосчётчика (величины объёма десорбата) регистрируют. При обработке результатов эксперимента вводят поправку на свободный объём системы. Результаты записывают в табл. 2.7.
Адсорбент - Объем адсорбента, л - Температура, °С - Барометрическое давление, мм рт.ст. - Газ - азот Таблица 2.7 Результаты измерений
По результатам измерений строят изотерму адсорбции a= f(P).
Вопросы для самоконтроля 1. Каким уравнением описываются изотермы адсорбции компонентов воздуха при комнатной температуре? 2. Какова методика волюмометрического определения изотерм адсорбции при повышенных давлениях? 3. Каковы особенности волюмометрического метода при измерениях при повышенном давлении?
Лабораторная работа 6 Определение изотермы адсорбции фенола из водных растворов на активном угле методом переменных концентраций Приборы и реактивы: − адсорбент – активный уголь; − адсорбтив – фенол; − бюксы стеклянные; − пробирки с пришлифованными пробками – 10 шт; − шейкер; − центрифуга; − спектрофотометр. Цель работы. Освоение метода переменных концентраций и определение изотермы адсорбции фенола из водных растворов на активном угле. Сущность работы. Метод измерения величины адсорбции в зависимости от относительной концентрации вещества в растворе заключается в определении разности концентраций адсорбтива в исходном и равновесном растворах. При использовании метода переменных концентрацийнесколько пробирок или колб (7-10 шт.) с навесками подготовленного адсорбента заливают растворами с различной концентрацией адсорбтива при соотношении Известно, что адсорбция на углеродных адсорбентах в значительной мере зависит от ряда факторов: концентрации адсорбтива, рН раствора, электронодонорных или электроноакцепторных свойств адсорбтива, пористости и поверхностных свойств адсорбента, которые связаны с присутствием окисленных функциональных групп и их поверхностного заряда. Для того чтобы получить надёжные данные по адсорбции фенола, необходимо фиксировать все параметры, которые могут оказывать влияние на процесс адсорбции. Порядок проведения работы Предварительные эксперименты: − определение времени установления равновесия в системе фенол − активный уголь; − калибровка спектрофотометра (методика проведения калибровки приведена ниже). Подготовка адсорбента Подготовка адсорбента заключается в очистке его поверхности от различных адсорбированных им паров и газов, а также от паров воды, которые могли быть поглощены из воздуха. Для этого образец активного угля в виде порошка прогревают в сушильном шкафу при температуре 100 оС. По достижении постоянной массы (примерно через 2 ч) бюкс с образцом вынимают из сушильного шкафа. После охлаждения бюкса в эксикаторе с цеолитом, бюкс с образцом взвешивают и определяют навеску угля. Навески адсорбента (10 шт.) взвешивают на аналитических весах (~ 0,1 г). Каждая навеска (m, г) загружается в заранее приготовленные колбы или пробирки, куда заливают растворы адсорбтива. Приготовление растворов Рассчитывают количество фенола, необходимое для приготовления раствора исходя из предельной растворимости. Предельную растворимость находят по справочным данным [17]. Растворимость фенола в воде при 18 оС составляет 0,88 моль/л. Рассчитанное количество фенола взвешивают на аналитических весах и в мерной колбе растворяют в дистиллированной воде, после чего доводят до метки и перемешивают до полного растворения вещества. Последующие растворы готовят методом разбавления исходного раствора. Начальные концентрации приготовленных растворов с 0 составляют от 5 до 30 ммоль/л. Результаты калибровки
По полученным данным строят калибровочный график зависимости lg T =f(c) (рис. 2.15), по которому определяют концентрацию исследуемых
Линеаризацию данных калибровки проводят по методу наименьших квадратов. Данные калибровки вносят в компьютер для автоматизации расчетов. Результаты опытов заносят в табл. 2.9. Таблица 2.9 Вопросы для самоконтроля 1. На чём основан метод определения изотерм адсорбции вещества из водных растворов? 2. Какие методы используются для определения концентрации органических веществ в водном растворе? 3. Какие адсорбенты применяют для очистки сточных вод от органических веществ?
Кинетика адсорбции Вопросы для самоконтроля 1. В чём суть волюмометрического метода измерении адсорбции газов? 2. В чём заключается предварительная подготовка адсорбента перед адсорбционными измерениями? 3. Как определяются кинетическая кривая, степень отработки адсорбционной ёмкости, величина равновесной емкости адсорбента, значение коэффициента внутренней диффузии?
ТЕРМИЧЕСКИЙ АНАЛИЗ Теоретическая часть Термический анализ является одним из важнейших методов физико-химического исследования веществ [22-26]. При воздействии высоких температур протекают химические реакции (дегидратация, разложение и т.д.) и физические превращения (плавление, полиморфные переходы и т.д.). Термический анализ широко используется при исследовании твёрдофазных процессов, реакций термического разложения и фазовых превращений веществ. В основе метода лежит зависимость физических или химических свойств материалов от температуры. В зависимости от измеряемого параметра различают основные методы исследований: – дилатометрия – измерение линейных и объемных размеров материалов; – дифференциально-термический анализ (ДТА) – определение разности температур между образцом и эталоном; – дифференциально-сканирующая калориметрия (ДСК) основана на непрерывной регистрации разности теплового потока от образца и эталона как функции температуры; – термогравиметрический анализ (ТГА) – определение величины и характера изменения массы образца в условиях программируемого подъема температуры; – синхронный термический анализ предполагает одновременное использование термогравиметрии и дифференциальной сканирующей калориметрии (ТГА/ДСК) или термогравиметрии и дифференциально-термического анализа (ТГА/ДТА). Наиболее широко применяются термогравиметрический и дифференциально-термический анализы. Термогравиметрический метод анализа основан на регистрации изменения массы образца при повышении температуры. Изменение массы твёрдых тел обусловлено протекающими физико-химическими процессами. Для процессов разложения и диссоциации характерно уменьшение массы образца, для реакции окисления, напротив − увеличение массы. Установка для термогравиметрических исследований состоит из термовесов, предназначенных для непрерывной регистрации изменений массы, печи, в которую помещен образец, термопары и программного регулятора температуры. Термогравиметрический анализ (ТГА) заключается в измерении зависимости массы твердого образца от температуры среды, в которую он помещен. Кривая зависимости потери массы от температуры (кривая ТГ, рис. 4.1) имеет вид плато, горизонтальный участок которой говорит об устойчивости химического соединения в данном интервале температур и отсутствии химических превращений (при этом физические превращения не исключаются), вертикальный участок на кривой свидетельствует о химическом разложении материала.
Рис. 4.1. Термогравиметрическая кривая (ТГ) разложения кристаллогидрата оксалата кальция
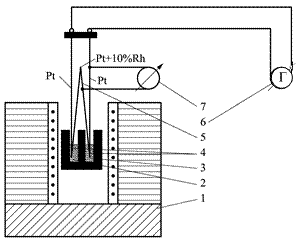
На основании кривой ТГ можно определить потерю массы при нагревании (Δ m), что позволяет оценить содержание примесей в анализируемом материале. Для этого необходимо знать состав химического соединения, относящегося к данному температурному плато, и состав продукта разложения. К сожалению, кривая ТГ не позволяет точно определить температуру разложения (истинную температуру химической реакции). Температуру химической реакции можно с высокой точностью определить, если воспользоваться дифференциальной формой записи, показывающей скорость изменения массы образца Возможны следующие варианты методов исследований: изотермический, когда масса образца изменяется на протяжении некоторого времени при выбранной температуре, квазистатический, когда образец нагревается при каждой из ряда возрастающих температур до достижения постоянного значения массы, и динамический, когда температура среды, окружаемой нагреваемый образец, изменяется по заданному закону. Из большого числа различных типов химических реакций наиболее хорошо изучены реакции разложения, на протекание которых в статических условиях в значительной степени влияют давление и состав газовой фазы, а в проточных системах – гидродинамические условия. Для определения состава газовой фазы используют химические и инструментальные методы анализа. Для этого должен быть предусмотрен непосредственный контакт ячейки с измерительным прибором (манометром или газовой бюреткой). Для определения количественного состава газовой фазы часто прибегают к вымораживанию, поглощению или превращению продуктов в легко анализируемую форму. Метод дифференциального термического анализа (ДТА) основан на сравнении термических свойств образца исследуемого вещества и термически инертного эталона, в качестве которого применяются вещества, не претерпевающие никаких изменений при нагревании до требуемой температуры. В качестве эталонов применяются прокаленные оксиды алюминия или магния. В данном случае регистрируемым параметром служит разность температур, измеряемая при нагревании или охлаждении образца и эталона с постоянной скоростью. ДТА-кривая получается с помощью дифференциальной термопары (рис. 4.2).
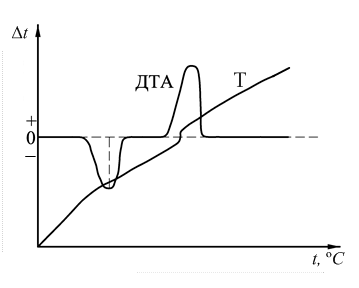
Рис. 4.2. Схема дифференциальной и простой термопар: 1– электропечь; 2 – исследуемое вещество; 3 – эталонное вещество; 4 – горячие концы дифференциальной термопары; 5– холодный спай дифференциальной термопары; 6 – гальванометр записи кривой ДТА; 7 – гальванометр записи температурной кривой. Дифференциальная термопара представляет собой две обычные термопары, соединенные между собой одноименными концами, образующими холодный спай. Два других конца подключены к прибору, позволяющему фиксировать изменения в цепи электродвижущей силы (ЭДС), возникающей при нагревании горячих спаев термопары. Один горячий спай помещён в исследуемое вещество, а другой в эталонное. Схематическое изображение простой (Т) и дифференциальной (ДТА) кривых нагревания приведено на рис. 4.3. Нулевой линией называется параллельная оси абсцисс линия, проведенная через начало хода кривой ДТА. Часто после завершения процесса разложения кривая ДТА не возвращается к нулевой линии, а идет параллельно ей или под некоторым углом. Эта линия хода термической кривой носит название базисной линии. Расхождение базисной и нулевой линий обусловлено различными теплофизическими свойствами исследуемого и эталонного веществ.
Рис. 4.3. Схематическое изображение простой (Т) и дифференциальной (ДТА) кривых нагревания
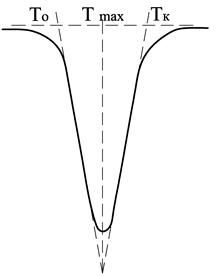
Экстремумы (пики) на кривой ДТА на базисной линии соответствуют термическим эффектам. Так, эндотермические превращения отображаются отклонением кривой ДТА ниже нулевой линии, экзотермические – выше нулевой линии. Первые обычно обозначаются знаком минус, вторые – плюс. Положение термических эффектов на термограмме кривой ДТА характеризуется температурными границами протекания реакции. Значение температур определяют графически, снося соответствующие точки с кривой ДТА на простую температурную кривую, а затем на ось температур. Устанавливают температуры начала Т 0, максимума Т max и окончания термического эффекта Тк (рис. 4.4). За начало процесса принимается температура Т 0, при которой кривая ДТА отклоняется от нулевой линии, фиксируя эндо- или экзотермический пик. При расшифровке результатов анализа часто указывают не начало и конец реакции, а интервал температур, в котором она протекает, т.е. Т 0 – Тк, или температуру максимума термического эффекта Т mах. Последнюю устанавливают как точку пересечения касательных к правой и левой ветвям пика.
Рис. 4.4. Определение температур начала, конца и максимума термического эффекта
Вследствие трудностей, связанных с графическим определением температуры начала реакции, наиболее надёжной характеристикой является температура Т mах. Изменения температуры образца вызываются физическими переходами или химическими реакциями, связанными с изменением энтальпии. К ним относятся фазовые переходы, плавление, перестройка кристаллической структуры, реакции дегидратации и разложения, разрушение кристаллической решётки и др. В общем случае фазовые переходы, дегидратация, восстановление и некоторые реакции разложения сопровождаются эндотермическими эффектами, а кристаллизация, окисление и отдельные процессы термолиза – экзотермическими эффектами.
Дериватография Дериватография (от лат. derivatus – отведённый, отклонённый и греч. grapho – пишу) –комплексный метод исследования химических и физико-химических процессов, происходящих в твёрдом теле в условиях программного подъема температуры, получивший в настоящее время широкое распространение. Дериватография основана на сочетании дифференциального термического анализа с другими физико-химическими методами: термогравиметрией, масс-спектрометрией и т.д. Дериватографом называется прибор, совмещающий термогравиметрический и дифференциальный термический анализы. При проведении дериватографических исследований регистрируются четыре кривые (рис. 4.5): – зависимость температуры от времени носит линейный характер и показывает скорость нагрева образца (Т); – термогравиметрическая кривая показывает зависимость изменения массы от температуры нагрева образца (ТГ); – кривая дифференциально-термического анализа отражает тепловые эффекты процессов, происходящих при нагревании вещества (ДТА); – дифференциально-термогравиметрическая кривая отражает скорость изменения массы образца (ДТГ). Основным отличием дериватографа от других термических установок является тождественность условий опыта, что достигается путем одновременной регистрации всех кривых на одном листе для одной и той же навески при соответствующих одинаковых температурах нагрева. Совместное получение простой и дифференциальной кривых даёт возможность обнаружить и количественно оценить даже очень слабые эффекты потери массы в изучаемом веществе.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-07-18; просмотров: 250; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.14.253.152 (0.163 с.) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
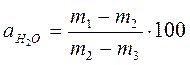 , (2.16)
, (2.16)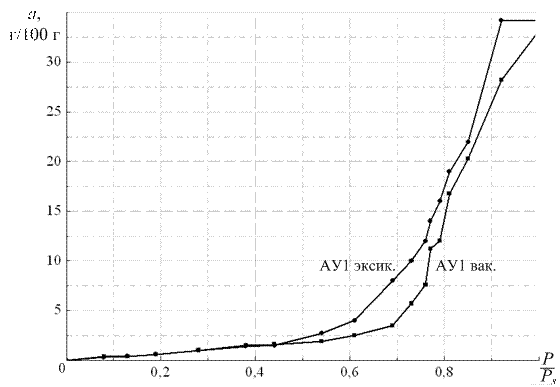
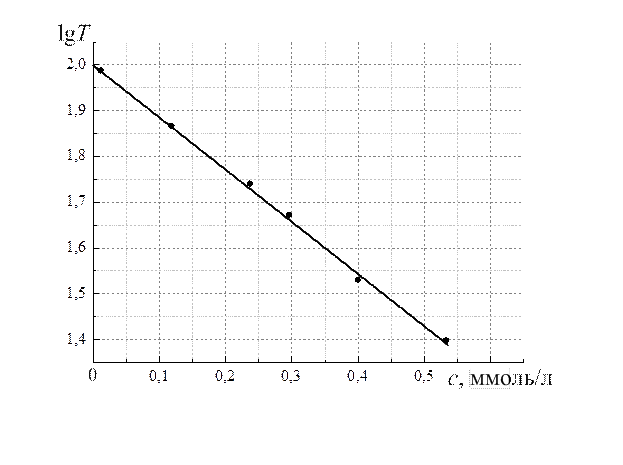 Рис. 2.15. Калибровочная кривая, связывающая степень пропускания и концентрацию водных растворов фенола при l =277 нм
Рис. 2.15. Калибровочная кривая, связывающая степень пропускания и концентрацию водных растворов фенола при l =277 нм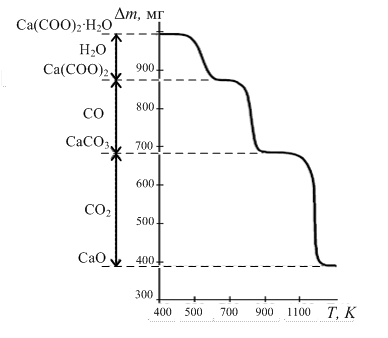
 от времени нагрева t. Каждый пик на дифференциальной кривой ДТГ соответствует максимальной скорости изменения массы, т.е. истинной температуре химической реакции.
от времени нагрева t. Каждый пик на дифференциальной кривой ДТГ соответствует максимальной скорости изменения массы, т.е. истинной температуре химической реакции.