
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Обзор теорий растворов неэлектролитовСодержание книги Поиск на нашем сайте Глава 6. Истинные растворы Для понимания растворов электролитов крайне важно выяснить, что представляет собой кинетическая частица, которую мы назвали ионом… Р. Робинсон, Р. Стокс. Растворы электролитов. Экспериментальной основой для создания общих положений теории растворов служат фундаментальные законы Вант-Гоффа, Рауля, Генри, а также результаты экспериментов Д.И. Менделеева, И.А. Каблукова, Дж.Г. Пойнтинга и других исследователей. Однако разработать единое учение о растворах, которое можно было бы использовать для описания свойств растворов электролитов и неэлектролитов, пока не удается. Тем не менее, на рис. 6.1 представлена схема, показывающая в первом приближении связи между основными учениями теории растворов.
Рис. 6.1. Взаимосвязи между важнейшими учениями теории растворов Из этой схемы, видно, что хотя теории растворов опираются на общую экспериментальную базу, их развитие идет по двум направлениям - отдельно для электролитов и неэлектролитов. По-видимому, причина этому в том, что Ф.М. Рауль, Дж. Генри, Я.Г. Вант-Гофф вывели свои законы, исследуя разбавленные системы, в случае которых не существует заметного влияния природы растворенных частиц на исследованные эффекты. Но это влияние возрастет с увеличением концентрации растворов, где в полной мере начинает проявляться взаимодействие частиц, предсказанное Д.И Менделеевым и Дж. Г. Пойнингом. Растворы неэлектролитов Структура раствора и природа Растворы электролитов Терминология, классификация и основные понятия К электролитам[3] относят вещества, молекулы которых при растворении или плавлении диссоциируют на ионы. Существуют различные классификации электролитов. Электролиты, молекулы которых распадаются на два иона, называются симметричными, или бинарными. Остальные электролиты считаются несимметричными. Дополнительно тип электролита устанавливают по заряду образующихся при диссоциации ионов. Если образуются однозарядные ионы, электролит относят к типу 1,1 – валентных электролитов (КCl, NaNO3), если двухзарядные – к типу 2,2 – валентных (CaSO4), например, К2SO4 относится к типу 1,2-, а CaCl2 - к типу 2,1 – валентных электролитов и т.п. Часто электролиты делят по способности молекул распадаться при растворении на ионы на сильные и слабые. Молекулы сильных электролитов диссоциируют на ионы полностью. Поэтому в растворах сильных электролитов присутствуют только молекулы растворителя и ионы растворенного вещества. Классическое равновесие между исходными молекулами и ионами в этих системах отсутствует. Примерами сильных электролитов в водных растворах могут быть щелочи (KOH), кислоты (HNO3, HCl), а также значительное число неорганических и ряда органических солей. Слабые электролиты лишь частично диссоциируют на ионы. Способность к диссоциации выражают с помощью понятия степень диссоциации (a). Величина a оценивается как доля продиссоциированных молекул из общего числа молекул, перешедших в раствор, т.е.
К слабым электролитам можно отнести большинство органических кислот, солей, оснований (HCN, CH3COONH4) и многие неорганические соединения, образующие в растворе многозарядные ионы. Некоторые авторы [11] оценивая степень диссоциации децимолярных растворов электролитов к сильным относят те, у которых a > 0,3, а к слабым - если a < 0,03. Электролиты, степень диссоциации которых лежит в пределах 0,03<a<0,3, относят к электролитам средней силы. Другой способ классификации электролитов предложил Дж. О. М. Бокрис. В соответствие с его классификацией электролиты делят на истинные электролиты или ионофоры и потенциальные электролиты или ионогены. Ионофоры в природе находятся в составе ионных кристаллов. Типичный ионофор - NaCl. При растворении в воде между поверхностными ионами ионофора и диполями воды возникает электростатическое (ион-дипольное взаимодействие). Вследствие этого ионные связи в кристалле ионофора ослабевают, и происходит диссоциация. Этот процесс может быть усилен возникновением водородных связей между водой и некоторыми анионами, образующими ионофор, если эти анионы содержат сильно электроотрицательные элементы. Ионогены вне раствора существуют в виде молекул газа или в конденсированном состоянии. Типичный ионоген - HCl. При растворении молекулы ионогенов взаимодействуют с водой, образуя гидратированные ионы. Взаимодействие HCl с водой можно представить следующим уравнением: HCl + 5H2O «H3O+ + [Cl(H2O)4]ˉ‾. Специфика равновесия в растворах слабых электролитов Диссоциация молекул электролита на ионы подчиняется известным законам химического равновесия, описанным в главе 4. Поэтому в растворах слабых электролитов устанавливается динамическое равновесие между молекулами электролита и ионами. Принципиальным моментом при анализе ионных равновесий в реальных растворах является необходимость всякий раз решать можно ли в конкретном случае в расчетах использовать аналитические концентрации вместо активностей. Ответ достаточно прост, если речь идет растворах слабых электролитов. Для них, как правило, использование при расчетах равновесий концентраций не приводит большим ошибкам, а концентрационные зависимости оказываются весьма значимыми. В общем случае равновесие в растворах слабых электролитов без учета сольватации можно представить схематично соотношением: MxAy = x M l + + y A m – (6.1) а также соответствующей константой равновесия (К с), называемой в этом случае концентрационной константой диссоциации (Кдис)
В частном случае 1,1 – валентного электролита МА, диссоциирующего в соответствии с уравнением MA = M + + A –, для раствора, аналитическая концентрация которого равна с о, константу диссоциации можно выразить уравнением Оствальда:
Из уравнения (6.2 а) следует, что чем больше Кдис, тем выше степень диссоциации. Поэтому величина Кдис может служить мерой силы электролита (способности его молекул) диссоциировать. Для умеренно слабых электролитов, например Н3РО4 (первая ступень), Са(ОН)2 значения К дис лежат в пределах от 10-2 до 10-4, для более слабых электролитов, например СН3СООН и NH4OH, величина К дис находится в пределах от 10-5 до 10-9. Если К дис<10-10 электролит относят к очень слабым [5]. Считается, что уравнение Оствальда[6] желательно применять при концентрациях электролита меньше 0,01 моль/л. Для описания равновесия в более концентрированных растворах оно не годится, и следует использовать термодинамические константы диссоциации К т, выраженные через активности компонентов:
В случае некоторых электролитов, когда a<<1 можно принять 1– a=1, тогда формула (6.2 а) упрощается и приобретает следующий вид: К дис = a2 с о. (6.3) Учитывая, что константа диссоциации - величина постоянная при данной температуре, на основании уравнения (6.3) можно заключить, что с уменьшением концентрации электролита (разбавлением раствора) степень диссоциации увеличивается. Важной характеристикой равновесия в растворах слабых электролитов является изотонический коэффициент (i) – величина, показывающая, во сколько раз увеличивается общее число частиц (ионов и молекул), в результате диссоциации. Его величину несложно связать со степенью диссоциации. При анализе соотношения (6.1) можно показать, что равновесные концентрации MxAy, x M l + и y A m – равны соответственно (1-a) с о, a x с о и a y с о. Отсюда следует, что:
Обозначая x + y через n, преобразуем соотношение (6.4) в известную формулу (1.6): i = 1 + (n- 1) a. Правило ионной силы Способ учесть всю гамму взаимодействий в растворе нашел Дж. Н. Льюис. Он сформулировал правило ионной силы: в разбавленных растворах коэффициент активности сильного электролита одинаков для всех растворов одной и той же ионной силы. Ионная сила [†] раствора электролита (I с) выражается через полусумму произведения концентрации (с i) каждого из присутствующих в растворе ионов на квадрат заряда zi этого иона, т.е.:
В соответствии с этим правиломактивность отдельных ионов можно рассчитать по формулам: а + = у + × с +; а - = у - × с -, (6.10) где: с + и с - - концентрации катиона и аниона, выраженные в моль/л,[8] а у + и у -- соответствующие этим ионам коэффициенты активности, которые можно рассчитать по формулам, включающим ионную силу раствора. Ниже приведена методика расчета ионной силы на примере раствора, содержащего 1М С6Н12О6,, 0,01 М СН3СООН, 0,10 М СН3СОOK, а также 0,20 М СаС12. Учитывается, что в общем случае концентрации отдельных ионов равны произведению аналитической концентрации электролита на число ионов образующихся при диссоциации молекулы, по уравнению: М l А m = l M m + + m A l -. Решение. Известно, что сахар С6Н12О6 на ионы не распадается, а диссоциация уксусной кислоты ничтожно мала. Поэтому можно принять, что в приведенных условиях ионная сила зависит только от концентрации ионов, получающихся при растворении СН3СОOK и СаС12: с (К+) = 0,1 М, с (С1-) =2·0,2=0,4 М, с (СН3СОО-) = 0,1 М, с (Са2+) =0,2 М. В итоге имеем I = 1/2 × (0,1×12 + 0,4×12 + 0,1×12+0,2×22) = 0,7 M. Вычисления усложняются в случаях, если необходимо учесть действительную степень диссоциации слабого электролита. Коэффициент активности (yi) для отдельного иона i врастворе ионная сила которого не превышает 0,1 можно найти по формуле Дебая и Хюккеля:
где A и B – константы, зависящие от свойств растворителя и температуры раствора. (для воды при 298 К они соответственно равны 0,509 и 0,328); z -заряд иона; I с - ионная сила раствора; d - параметр, соответствующий диаметру гидратированного иона (табл.6.1). Таблица 6.1. Значения параметра d для различных ионов [31]
Формула (6.11) для расчета коэффициента активности отдельного иона имеет сугубо теоретическую ценность, и ее проблематично использовать для практических расчетов, поскольку в растворе одновременно присутствуют разнозаряженные ионы растворенного вещества. Поэтому вводят понятия средней ионной активности и среднего коэффициента активности. Среднюю активность электролита а ± оценивают исходя из общей активности электролита
где l и m - соответственно число положительных и отрицательных ионов, Подставляя выражения для активностей отдельных ионов из (6.9) в (6.12) получим a ± = = у ± с ±; a = a ±( l + m ), где у ± и × с ± - средний ионный коэффициент активности[9] и средняя ионная концентрация соответственно. В этом случае формулу Дебая и Хюккеля (6.11) следует переписать следующим образом:
Формула Дебая и Хюккеля дает удовлетворительные результаты для растворов ионной силы до 0,1М. Для интервала от 0,1 М до 1 М расчет часто выполняют по формуле Дэви
В случае разбавленных растворов можно принять, что lg y ±= Формула (6.15) представляет предельный закон Дебая и Хюккеля. Ее часто используют для грубых оценок коэффициентов активности. Контрольное задание 21 Выберите и обоснуйте правильное утверждение. 21.1. При смешивании равных объемов 2 моль/л растворов сахара и хлористого калия значение ионной силы раствора становится равным (моль/л): а) 0,5; б) 1,0; в) 2,0. 21.2. Увеличение ионной силы раствора электролита способствует образованию: а) сольватов; б) ассоциатов; в) не влияет на характер взаимодействий в растворе. Гидролиз солей
Гидролиз электролитов[10] можно определить как взаимодействие с водой ионов растворяемой соли с образованием слабодиссоциирующей кислоты или основания, а иногда слабодиссоц иирующих кислоты и основания одновременно. Важнейшим следствием гидролиза по одному из ионов соли является изменение рН раствора. Этот признак может отсутствовать при одновременном гидролизе по аниону и катиону. Поэтому его нельзя считать достаточным для доказательства отсутствия гидролиза. Другой характерный признак гидролиза - сжатие раствора, которое вызывается уплотнением растворителя вокруг ионов, подвергающихся гидролизу и приводит к существенному изменению структуры растворителя, а нередко и самих ионов, Механизм гидролиза включает две стадии: 1) диссоциацию молекул растворяемого электролита на ионы; 2) взаимодействие ионов с водой (протолиз). Первая стадия по сути представляет собой растворение вещества в воде. Эта стадия описана ранее. Здесь же акцент будет сделан на стадии протолиза, которая определяет характер гидролиза в целом. Без учета эффекта гидратации для бинарной соли 1,1 – валентного электролита типа МеА отдельные стадии гидролиза можно для различных случаев записать следующим образом: 1) диссоциация молекул электролита: МеА = Ме+ + Аˉ; 2) протолиз (взаимодействие ионов с водой). Таблица 6.2 Реакция |
Константа | рН | |||||||||||||||
| равновесия, Кр | гидролиза, К г | |||||||||||||||||
|
Гидролиз по аниону | ||||||||||||||||||
| Аˉ+ Н2О = НА + ОНˉ |  (1) (1)
| 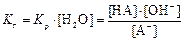 (2) (2)
| > 7 | |||||||||||||||
| Гидролиз по катиону* | ||||||||||||||||||
| Ме+ + Н2О = МеОН + Н+ |  (3) (3)
| 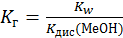 (4) (4)
| < 7 | |||||||||||||||
|
Характеристик растворов Под буферированием понимают поддерживание на заданном уровне тех или иных характеристик раствора. Такими характеристиками часто оказываются рН, концентрация ионов определенного вида, ионная сила и др. Буферирование выполняют, вводя в исследуемый раствор соответствующие буферные смеси или растворы отдельных солей в концентрациях достаточных для поддерживания на одном уровне выбранных характеристик. Буферная смесь (буфер) – смесь растворов веществ, часто электролитов, подобранных таким образом, чтобы можно было обеспечить при изменении состава среды постоянство заданных характеристик исследуемого раствора. С учетом задач исследователя состав буфера в каждом конкретном случае подбирается специально. Буфер рН. Буферные растворы для поддерживания заданных значений рН готовятся на основе сопряженных пар типа: слабая кислота - сильная соль этой кислоты и слабое основание – сильная соль этого основания. Буферирующее действие обусловлено тем, что добавляемые к буферу ионы водорода или гидроксида связываются компонентами буфера, не вызывая заметного смещения ионного равновесия в пределах некоторого достаточно узкого диапазона рН. Для различных буферных смесей диапазон буферирования, как правило, находится в пределах р К ±1 (р К - отрицательный логарифм константы диссоциации слабой кислоты или слабого основания). Меняя концентрации компонентов буфера, получают растворы с заданными значениями рН в пределах области буферирования. Исключение составляют универсальные буферные смеси с расширенными областями буферирования, в случае которых требуемые рН создаются добавками к основному составу буфера 0,2 М раствора NaOH. Составы некоторых буферных смесей приведены в табл. 6.3. Таблица 6.3 Составы и характеристики буферных смесей [34] Авторы |
Кислота |
Основание |
Интервал рН | |||||||||||||||
| Состав | р К (при 298 К) | |||||||||||||||||
| Двухкомпонентные смеси | ||||||||||||||||||
| С.П.Серенсен | НCl | - | Глицин | 1,0 …3,7 | ||||||||||||||
| Х.Т.С.Бриттон | Лимонная – C3H4OH(COOH)3 | pK (H4L)=3,1 pK (H3L-)=4,4 | NaOH | 2,2 … 6,5 | ||||||||||||||
| Т.С.Макливен | Муравьиная - HCOOH | 3,8 | NaOH | 2,8 … 4,6 | ||||||||||||||
| Х.Т.С.Бриттон - | Уксусная - CH3COOH | 4,76 | CH3COONa | 3,7 … 5,6 | ||||||||||||||
| В.М. Кларк | KH2PO4 | 7,21 | NaOH | 5,8 … 8,0 | ||||||||||||||
| В.М. Кларк | Борная - H3BO3 | 9,2 | NaOH | 8,0 … 10,0 | ||||||||||||||
| С.П. Серенсен | Глицин (аминоуксусная) – H2NCH2COOH | pK (СООН)=2,34 pK (NН2)=9,6 | NaOH | 8,2 … 10,1 | ||||||||||||||
| С.П. Серенсен | Глицин + Na2HPO4 | - | NaOH | 8,3 … 11,9 | ||||||||||||||
| И.М. Кольтгофф | Na2B4O7 | - | Na2CO3 | 9,2 … 11,0 | ||||||||||||||
| Универсальная буферная смесь | ||||||||||||||||||
| Х.Т.С. Бриттон –Р.А.Робинсон | 0,04 М раствор фосфорной, уксусной и борной кислот | 0,2 М NaOH | 1,8 … 11,9 | |||||||||||||||

 .
. . (6.2)
. (6.2) . (6.2 а)
. (6.2 а) . (6.2 б)
. (6.2 б)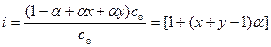 . (6.4)
. (6.4)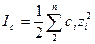 . (6.9)
. (6.9)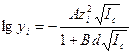 , (6.11)
, (6.11)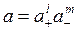 по формуле:
по формуле: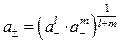 , (6.12)
, (6.12)  . (6.13)
. (6.13) (6.14)
(6.14) . Поэтому формула Дебая и Хюккеля становится проще. Учитывая (например для бинарных 1-1 – электролитов), что Ic = c,можно получить:
. Поэтому формула Дебая и Хюккеля становится проще. Учитывая (например для бинарных 1-1 – электролитов), что Ic = c,можно получить: (6.15)
(6.15) Оценку рН буферных растворов выполняют на основе анализа ионных равновесий, характеризующих данный буфер. Например, для двухкомпонентной буферной смеси слабой кислоты НА и ее соли NaA ионные равновесия можно представить набором уравнений, включающих закон действующих масс, а также условия электронейтральности и массового баланса:
Оценку рН буферных растворов выполняют на основе анализа ионных равновесий, характеризующих данный буфер. Например, для двухкомпонентной буферной смеси слабой кислоты НА и ее соли NaA ионные равновесия можно представить набором уравнений, включающих закон действующих масс, а также условия электронейтральности и массового баланса:
[H+] [A-] = K НА [HA];
[H+] [OH-] = Kw;
[H+] +[Na+] = [OH-] + [A-]; (6.17)
[HA] + [A-] = c HA + c NaА;
[Na+] = c NaA).
Сочетая эти уравнения при условии, что величины [H+] и [OH-] пренебрежимо малы по сравнению с аналитическими концентрациями кислоты и соли c HA и c NaA, а также допуская, что [A-] = c NaA и [HA] = c HA можно получить следующую простую формулу для расчета рН буфера (формула Хендерсона):
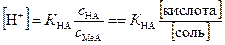 . (6.18)
. (6.18)
Подобным же образом нетрудно вывести выражение для оценки рН смеси слабого основания и его соли.
При использовании уравнения (6.18) необходимо иметь в виду, что оно не выполняется если:
· кислота является слишком сильной - нельзя пренебречь величиной [H+], или слишком слабой – нельзя пренебречь [OH-].
· аналитические концентрации c HA и c NaA слишком малы (по грубым оценкам меньше 10-3 моль/л).
Эффективность буферирования характеризуют буферной емкостью[12] (b) равной:
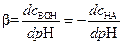
где: сМеОН и с НА - концентрация сильной щелочи или кислоты, специально добавляемых к буферному раствору.
Буферная емкость кислотно-основного буфера соответствует числу эквивалентов сильной кислоты или щелочи, которое необходимо добавить к одному литру раствора, чтобы изменить его рН на единицу. Величина b возрастает при увеличении начальных концентраций образующих буфер компонентов и максимальна приих равенстве. Из формулы (6.18) следует, при изменении соотношения между компонентами буфера в 10 раз (например, от значения 1:1 до значения 1:10) рН изменится на единицу. Этот интервал изменения отношения концентраций принимают за максимально возможный для буферных смесей. Смесь теряет свойства буфера при более высоких соотношениях концентраций составляющих компонентов.
Для двухкомпонентной буферной смеси буферную емкость можно рассчитать по уравнению:
 (6.19)
(6.19)
Типичным буфером рН является ацетатный буфер, Бриттона - Вальполя (см. табл.6.4). Этот буфер представляет собой смесь уксусной кислоты и ацетата натрия, концентрации которых задают соответственно требуемому значению рН в пределах рабочего диапазона рН (см. табл. 6.4).
Таблица 6.4
Вопросы
1. Каковы причины образования ассоциатов в растворах?
2. Как давление влияет на растворимость газа в жидкости?
3. Для приготовления литра 40% этанола следует взять 444 см3 90% раствора этанола и 581 см3 воды. Объясните, почему формально суммированный объем реагентов превышает 1000 см3?
4. Определите энтальпию растворения тростникового сахара в воде по данным для растворимости в 100 г воды при разных температурах
| Температура | 0 o C | 20 o C | 100 o C |
| Растворимость г на 100 г воды | 64,45 | 66,72 | 82,65 |
5. Две одноосновные органические кислоты при одинаковой молярной концентрации имеют степени диссоциации: первая -0,2, вторая -0,5. Константа диссоциации какой кислоты больше и во сколько раз?
6. Что означает термин "коллигативные свойства"? В чем проявляются эти свойства?
7. Давление пара водного раствора, содержащего нелетучее растворенное вещество, на 2 % ниже давления пара чистой воды. Определите моляльную концентрацию растворенного вещества.
8. Определите температуру кипения 2,0 % водного раствора глюкозы. Эбуллиоскопическая постоянная для воды - 0,512.
9. При температуре 27оС осмотическое давление раствора сахара в воде равно 1,0133·105 Па. Определите осмотическое давление этого раствора при 0 оС. Рассчитайте навеску, хлорида кальция которую необходимо взять, чтобы приготовить 100 см3 водного раствора изотоничного с этим раствором сахара при 0оС.
10. Сколько глицерина должно быть растворено (в %), чтобы давление пара раствора было на 2% ниже давления пара воды?
11. Выполните анализ единой схемы электролитической диссоциации, учитывающей явления сольватации и ассоциации.
12. Чем "ионогены" отличаются от “ионофоров”? Приведите механизм растворения ионофоров и ионогенов в воде.
13. Что такое ионная атмосфера? Какие факторы определяют радиус ионной атмосферы?
14. Приведите основные положения теории электролитов Аррениуса. В чем основные недостатки этой теории?
15. В чем состоит правило ионной силы? Приведите основные формулы для расчета ионной силы.
16. Приведите сущность явления осмос. Как можно рассчитать осмотическое давление растворов электролитов и неэлектролитов?
17. Рассчитайте осмотическое давление молока при нормальных условиях, если известно, что его температура замерзания в среднем равна -0,54 оС.
18. Кажущаяся степень диссоциации раствора, состоящего из 5,0 г хлористого натрия в 100 г воды равна 0,8. Рассчитайте температуру замерзания раствора (К к=1,86 °С)?
19. Рассчитайте навеску соли, которую необходимо взять, чтобы приготовить раствор заданной аналитической концентрации. Плотность растворов примите равной 1 г/см3. Формулу соли, объем раствора и аналитическую концентрацию, соответствующую вашему варианту см в табл.
Таблица
Исходные данные к расчетам навески соли для приготовления раствора
| Вариант | Формула соли | Объем раствора, см3 | Концентрация |
| 1 | CuSO4´5H2O | 200 | 10 % |
| 2 | Al2(SO4)3, | 200 | 0,1 н |
| 3 | KClO4 | 100 | 5% |
Оцените возможность гидролиза в растворе соли, соответствующей вашему варианту. Приведите уравнения гидролиза и оцените рН раствора.
- Если гидролиз в растворе соли заданного варианта не идет предложите формулу соли, подвергающейся гидролизу, и обоснуйте выбор.
- Рассчитайте ионную силу смесей, полученных смешиванием в равных объемах растворов, приведенных в таблице. Смешиваются два соседних раствора. Вариант отвечает раствору с меньшим номером.
- С учетом найденного значения ионной силы рассчитайте значения коэффициентов активности для ионов соли заданного варианта. Обоснуйте выбор использованного для расчета уравнения.
[*] Здесь и в дальнейшем предполагается, что ионы в растворе сольватированы.
[†] Следует помнить, что при расчетах ионной силы нейтральные частицы не учитывается, поскольку они не влияют на величину электростатических взаимодействий.
[1] Д. И. Менделеев на основе исследований плотности спиртовых растворов одним из первых пришел к выводу, о наличии взаимодействия между растворителем и растворяемым веществом.
[2] Такую сольватацию называют универсальной сольватацией и уподобляют процессу конденсации, т. е. переходу вещества из парообразного состояния в жидкое. В то же время допускается рассматривать конденсацию как автосольватацию самих молекул растворителя.
Нередко вводят понятия идеальной и реальной сольватации. В таком случае акцент делается на характере структуры основной массы растворителя. Принимается, что эта структура при идеальной сольватации не нарушена и раствор обладает свойствами бесконечно разбавленного раствора. При реальной сольватации взаимодействие сольватированных частиц с молекулами растворителя существенно меняет данную структуру.
[3] Иногда электролитами называют жидкие или твердые вещества и системы, способные проводить электрический ток с помощью присутвтвующих в них ионов.
[4] На это обстоятельство обращал внимание Д.И. Менделеев. Он замечал, что с принятием представлений о диссоциации Аррениуса "становится необходимым принять в растворе свободные ионы, подобные атомам Cl или Na, происшедшими без видимой затраты энергии, необходимой для их разъединения".
[5] Термин "сольватация" был введен И.А. Каблуковым в 1891 году, чтобы обозначить взаимодействия,которые возникают между растворенным веществом и растворителем.
[6] Уравнение (6.2) представляет закон разведения Оствальда. Это уравнение получают, учитывая, что [M+] = [A-] = acо, а [MA] = (1- a)cо.
Следует иметь в виду, что квадратные скобки - символ только равновесной молярной концентрации некоторого вещества В (моль/л). Другие выражения концентраций или неравновесные молярные концентрации обозначать этим символом не рекомендуется.
[7] Образование ассоциатов в значительной мере характерно для неводных растворителей. Но и в случае водных растворов наличие ассоциатов является доказанным фактом. В частности, установлено наличие таких образований как BaCl+, AgCl2-, LiCl2-.
[8] Следует отметить, что в зависимости от единиц измерения концентрации меняется и обозначение, и численная величина коэффициента активности и ионной силы. См. табл.
| Аналитическая концентрация, единицы измерения | Обозначение | |||
| концентрации | коэффициента активности | активности | ионной силы | |
| Молярная, моль/л | c i | y i | a i | I c |
| Моляльная, моль/кг | c m,i | gi | am, i | I m |
| Мольные доли (безразм.) | N i | f | ||
[9] Средний ионный коэффициент активности электролита y± равен y± = (y+ l y- m)1/ l+ m. Средняя ионная концентрация с ± равна с ± = (с + l с- m)1/ l+ m.
[10] Наряду с гидролизом солей этим термином обозначают и взаимодействие с водой других классов химических веществ: углеводов, белков, эфиров, бинарных соединений, в числе которых карбиды, нитриды, фосфиды и пр. Поэтому нередко гидролиз определяют, как обменное взаимодействие молекул растворенного соединения с водой. В этом случае как бы признается, что при растворении вещество переходит в раствор в молекулярном виде, что в случае электролитов как было показано выше не так.
[11] Константу гидролиза в литературе иногда путают с константой основности. Для протолитического равновесия типа А-+ Н2О = Н2О+ ОН- константа основности имеет вид 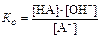 .
.
[12] Термин буферная емкость (buffer index) ввел Д.Д. ван Слайк в 1922 г. Иногда ее определяют как обратный наклон, имея ввиду наклон кривой титрования равный d pH / dc.
[13] Иногда пользуются записью уравнения закона Фика (6.25), в которой градиент концентрации заменяют на градиент плотности диффундирующего вещества, т.е.:  где m i - масса диффундирующего вещества, r - плотность вещества i.
где m i - масса диффундирующего вещества, r - плотность вещества i.
[14] Ярким примером проявления осмоса в природе служит тургор – перемещение воды через мембраны растительных клеток. Упругость зеленой массы растений в первую очередь связывают именно с этим явлением. В пищевых технологиях осмос проявляется при консервировании продуктов в концентрированных солевых или сахарных средах. В таких средах микроорганизмы гибнут вследствие обезвоживания, которое вызывают силы осмотического давления..
[15] Следует отметить, что закон Вант-Гоффа и понятие осмотического давления имеют смысл только в отношении очень разбавленных солевых растворов и коллоидных систем. В случае растворов полимеров (в том числе природных, типа полисахаридов или белков) часто пользуются выражением для осмотического давления, содержащим вириальные осмотические коэффициенты: p= RT (c 2M2 + A2 c 22 + A3 c 23 + ….)
где М2 - молекулярная масса полимера, А2, А3 - вириальные коэффициенты, зависящие от формы молекул и характера межмолеулярных сил, действующих между этими молекулами. Для полисахаридов в водных растворах 0,15 -0,2 М соли значения второго вирального коэффициента равны примерно 2,3 ∙10-3 моль∙см3/г2 [Биохимическая термодинамика. Под ред. М.Джоунс. Пер с англ. "Мир", М. 1982, с 196-197.
Глава 6. Истинные растворы
Для понимания растворов электролитов крайне важно выяснить, что представляет собой кинетическая частица, которую мы назвали ионом…
Р. Робинсон, Р. Стокс. Растворы электролитов.
Экспериментальной основой для создания общих положений теории растворов служат фундаментальные законы Вант-Гоффа, Рауля, Генри, а также результаты экспериментов Д.И. Менделеева, И.А. Каблукова, Дж.Г. Пойнтинга и других исследователей. Однако разработать единое учение о растворах, которое можно было бы использовать для описания свойств растворов электролитов и неэлектролитов, пока не удается. Тем не менее, на рис. 6.1 представлена схема, показывающая в первом приближении связи между основными учениями теории растворов.

Рис. 6.1. Взаимосвязи между важнейшими учениями теории растворов
Из этой схемы, видно, что хотя теории растворов опираются на общую экспериментальную базу, их развитие идет по двум направлениям - отдельно для электролитов и неэлектролитов. По-видимому, причина этому в том, что Ф.М. Рауль, Дж. Генри, Я.Г. Вант-Гофф вывели свои законы, исследуя разбавленные системы, в случае которых не существует заметного влияния природы растворенных частиц на исследованные эффекты. Но это влияние возрастет с увеличением концентрации растворов, где в полной мере начинает проявляться взаимодействие частиц, предсказанное Д.И Менделеевым и Дж. Г. Пойнингом.
Растворы неэлектролитов
Обзор теорий растворов неэлектролитов
Главная задача известных теорий растворов - предсказание свойств растворов по составу и характеристикам чистых компонентов. Между тем строение, структура и свойства растворов зависят в основном от концентрации растворенного вещества, природы компонентов раствора, характера и интенсивности их взаимодействий, т.е. определяются, прежде всего, механизмом растворения.
Механизм растворения различных веществ системно изучает химическая теория растворов, которая утверждает, что взаимодействия в растворах могут происходить как на молекулярном, так и на ионном или ионно-молекулярном уровнях. У истоков химической теории для растворов неэлектролитов стояли Д.И. Менделеев, И.А. Каблуков, Дж. Г. Пойнтинг и др.
Чтобы отразить такие взаимодействия, И.А. Каблуков в 1891 году ввел термин сольватация и предположил, что диссоциация происходит благодаря сольватации, продуктами которой являются сольват - непрочные соединения, образованные молекулами растворяемого вещества и растворителя. Дж.Г. Пойнтинг в 1896 году выдвинул гипотезу о том, что нелетучий компонент понижает давление пара растворителя в результате взаимодействия молекул этого компонента и растворителя. Д.И. Менделеев, обобщив результаты собственных исследований[1] и выводы других авторов, сформулировал принципиальные положения этой теории, а именно:
· причиной образования и существования раствора является взаимодействие исходных компонентов (растворителя и растворенных веществ) с образованием неустойчивых химических соединений, которые частично диссоциируют;
· все компоненты раствора находятся
|
| Поделиться: |



