
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Активность и коэффициент активности электролитов. Ионная сила раствора. Распределение ионов в раствореСодержание книги
Похожие статьи вашей тематики
Поиск на нашем сайте Активность растворенной соли а может быть определена по давлению пара, температуре затвердевания, по данным о растворимости, методом ЭДС. Все методы определения активности соли приводят к величине, характеризующей реальные термодинамические свойства растворенной соли в целом, независимо от того, диссоциирована она или нет. Но термодинамические свойства различных ионов не могут быть определены порознь из опытных данных без дополнительных допущений; мы можем измерить только средние термодинамические величины для ионов, на которые распадается молекула этого вещества. Пусть диссоциация соли происходит по уравнению Аn+ Вn- = n+ Аz+ + n- Bz-. Так как нет методов экспериментального определения значений а+ и а –в отдельности, то вводят среднюю ионную активность а ±, определяемую соотношением а ±n = а. Таким образом, мы имеем две величины, характеризующие активность растворенной соли. Первая из них - это мольная активность, то есть активность соли, определяемая независимо от диссоциации; она находится теми же экспериментальными методами и по тем же формулам, что и активность компонентов в неэлектролитах. Вторая величина - средняя ионная активность а ±. Введем теперь коэффициенты активности ионов g+ и g–, среднюю ионную моляльность m ± и средний ионный коэффициент активности g±: a + =g+ m +, a – =g– m –, a ± = g± m ±, где g± = (g+n+×g-n-)1/n, m ± = (m +n+× m -n-)1/n = (n+n+×n-n-)1/n m. Величина g± является важной характеристикой отклонения раствора соли от идеального состояния. В растворах-электролитах, как и в растворах-неэлектролитах, могут быть использованы следующие активности и коэффициенты активности: g± = f ± = Основными методами измерения величин коэффициентов активности являются криоскопический и метод ЭДС. Ионной силой I (или ионной крепостью) раствора называется полусумма произведений концентрации каждого иона на квадрат числа его заряда (валентности), взятая для всех ионов данного раствора. Если использовать моляльность как меру концентрации, то ионная сила раствора определяется выражением I = где i - индексы ионов всех солей в растворе; mi = n i m. Льюис и Рендалл открыли эмпирический закон ионной силы: средний ионный коэффициент активности g± диссоциирующего на ионы вещества является универсальной функцией ионной силы раствора, то есть в растворе с данной ионной силой все диссоциирующие на ионы вещества имеют коэффициенты активности, не зависящие от природы и концентрации данного вещества, но зависящие от числа и валентности его ионов. Закон ионной силы отражает суммарное взаимодействие ионов раствора с учетом их валентности. Этот закон точен лишь при очень малых концентрациях (m ≤ 0,01); уже при умеренных концентрациях он верен лишь приблизительно. В соответствии с этим законом, в разбавленных растворах сильных электролитов lg g± = - А
2.3) ТЕОРИЯ ЭЛЕКТРОЛИТОВ ДЕБАЯ И ГЮККЕЛЯ. Вокруг каждого отдельного иона существует ионная атмосфера (ионное облако) - сфера, состоящая из ионов противоположного знака. Ионы, входящие в состав сферы, непрерывно обмениваются местами с другими ионами. Все ионы раствора равноценны, каждый из них окружен ионной атмосферой, и в то же время каждый центральный ион входит в состав ионной атмосферы какого-либо другого иона. Существование ионных атмосфер и есть тот характерный признак, который, по Дебаю и Гюккелю, отличает реальные растворы электролитов от идеальных. ™ Ионы стремятся к сближению с ионами противоположного знака ™ Раствор в целом электронейтрален, но вблизи катионов находится избыток анионов и наоборот ™ Окружение центрального иона называется ионной атмосферой ™ Взаимодействие с ионной атмосферой понижает энергию Гиббса иона rD – дебаевский радиус ионной атмосферы.
При разработке этой теории были сделаны следующие допущения: 1. Число ионов в электролите можно определить из аналитической концентрации электролита, т.к. он считается полностью диссоциированным (a = 1). Теорию Дебая и Гюккеля поэтому иногда называют теорией полной диссоциации. Однако ее можно применить и в тех случаях, когда a ¹ 1. 2. Распределение ионов вокруг любого центрального иона подчиняется классической статистике Максвелла-Больцмана. 3. Собственными размерами ионов можно пренебречь по сравнению с расстояниями между ними и с общим объемом раствора. Т.о., ионы отождествляются с материальными точками, и все их свойства сводятся лишь к величине заряда. Это допущение справедливо только для разбавленных растворов. 4. Взаимодействие между ионами исчерпывается кулоновскими силами. Наложение сил теплового движения приводит к такому распределению ионов в растворе, для которого характерна статистическая шаровая ионная атмосфера. Это допущение справедливо лишь для разбавленных растворов. При повышении концентрации среднее расстояние между ионами уменьшается, и наряду с электростатическими силами появляются другие силы, действующие на более близком расстоянии, в первую очередь силы Ван-дер-Ваальса. 5. При расчетах принимается, что диэлектрические проницаемости раствора и чистого растворителя равны; это справедливо только в случае разбавленных растворов. Т.о., все допущения Дебая и Гюккеля приводят к тому, что их теория может быть применима только к разбавленным растворам электролитов с ионами низкой валентности. Первое приближение (предельный закон Д-Х): lg f ± = - A |z+×z-|
Предельный закон Дебая-Гюккеля дает верные значения коэффициентов активности 1-1 зарядного электролита, особенно в очень разбавленных растворах. Во втором приближении они отказались от представления об ионах как о материальных точках (допущение 3) и попытались учесть конечные размеры ионов, наделив каждый электролит некоторым средним диаметром а (при этом изменяется и допущение 4). Во втором приближении средний коэффициент активности описывается уравнением: lg f ± = - где А сохраняет прежнее значение; а условно названо средним эффективным диаметром ионов, имеет размерность длины, фактически - эмпирическая постоянная; В = c/ Гюккель учел уменьшение диэлектрической проницаемости с ростом концентрации растворов. Ее уменьшение вызывается ориентацией диполей растворителя вокруг иона, в результате чего снижается их реакция на эффект внешнего поля. Уравнение Гюккеля выглядит следующим образом: lg f ± = - где С - эмпирическая константа. При удачном подборе значений В и С формула Гюккеля хорошо согласуется с опытом и широко используется при расчетах. Дальнейшее развитие: ™ Учет ионной ассоциации ™ Учет взаимодействия молекул растворителя с ионами В процессе развития теории Дебая-Гюккеля и последовательного отказа от принятых допущений улучшается сходимость с опытом и расширяется область ее применимости, однако это достигается ценой превращения теоретических уравнений в полуэмпирические. Достоинства и недостатки: Ограниченность Дебая - Хюккеля теории обусловлена пренебрежением ассоциаций ионов, представлением о р-рителе как о непрерывной среде, характеризуемой только значением e, т. е. неучетом мол. структуры р-рителя и его взаимод. с ионами. Дебая - Хюккеля теория является основой теории электропроводности разбавл. р-ров сильных электролитов, разработанной Л. Онсагером. Она позволяет объяснить увеличение электропроводности р-ра при повышении напряженности постоянного электрич. поля (эффект Вина) и в высокочастотном поле (эффект Дебая-Фалькенхагена). В этих условиях ионная атмосфера, тормозящая движение ионов, не успевает образоваться (см. Электропроводность электролитов).
|
||
|
|
Последнее изменение этой страницы: 2016-08-14; просмотров: 1239; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.41 (0.01 с.) |

 - практический коэффициент активности (средний моляльный);
- практический коэффициент активности (средний моляльный); - средний мольный коэффициент активности.
- средний мольный коэффициент активности. ,
, .
.

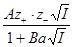 (2)
(2)


