Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Московский авиационный институт. Московский авиационный институтСтр 1 из 28Следующая ⇒
МОСКОВСКИЙ АВИАЦИОННЫЙ ИНСТИТУТ (ГОСУДАРСТВЕННЫЙ ТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ) ____________________________________________________ РАСТВОРЫ. ЭЛЕКТРОХИМИЯ Учебное пособие к лабораторному практикуму по химии Утверждено на заседании редсовета 17 октября 2007 г.
М О С К В А Издательство МАИ−ПРИНТ
2 0 0 9 Авторы: Н.А. Бункина, Н.С. Буданова, В.Н. Гразлов, С. И. Селиванова, М. А. Семенова, В. А. Новожилов, А. А. Фармаковская, Н. П. Жарова, Г. Н. Устюжанинова. РАСТВОРЫ. ЭЛЕКТРОХИМИЯ Учебное пособие к лабораторному практикуму по химии/ / Под редакцией Н.С. Будановой и А.А. Фармаковской – М.: Изд-во МАИ-ПРИНТ, 2009, с:, ил.
УДК:54+541.8+541.13] (076)
Пособие содержит теоретическое введение и описание лабораторных работ по двум основным разделам курса химии: «Растворы» и «Электрохимия». В разделе «Растворы» рассмотрены вопросы равновесия в растворах неэлектролитов и электролитов, буферные растворы, коллоидные растворы, адсорбционное равновесие, ионообменная хроматография. Раздел «Электрохимия» посвящен вопросам, связанным с теорией гальванических элементов, электролиза, коррозии, а также практическим применениям электрохимических методов анализа. Пособие предназначено для студентов 1-го курса всех технических факультетов МАИ дневной и вечерней форм обучения.
Рецензенты: кафедра физики и физической химии Военно-инженерной академии им. Н.Е. Жуковского (зав. кафедрой профессор В.В. Чернышев); д-р х. н., профессор Н.Н. Желиговская. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ РАЗДЕЛ 1. РАСТВОРЫ
В окружающем нас мире практически все жидкости – это растворы. Растворы (жидкие, твердые или газообразные) представляют собой гомогенную (однородную) систему из двух или большего числа веществ (компонентов), равномерно распределенных друг в друге. При растворении одного компонента в другом или смеси других происходит изменение качественного и количественного состава растворов. Поэтому кратко раствор можно определить как многокомпонентную гомогенную систему переменного состава. Составными частями раствора являются растворитель (вещество, которое находится в избытке по сравнению с остальными веществами) и растворенные вещества. По концентрации растворенного вещества растворы принято делить на ненасыщенные, насыщенные и пересыщенные.
Для характеристики количественного состава раствора при химических экспериментах чаще всего используются следующие концентрации: 1) массовые проценты — число массовых частей растворенного вещества в 100 массовых частях раствора, %; 2) молярность М — число молей растворенного вещества в 1 л раствора [г-моль/л]; 3) нормальность N — число грамм-эквивалентов растворенного вещества в 1 л раствора [г-экв/л]; 4) моляльность 5) титр Т — число граммов растворенного вещества в 1 см3 (мл) раствора [г/см3]; 6) мольная доля — отношение числа молей растворенного вещества к сумме числа молей всех компонентов раствора
Глава 1. РАВНОВЕСИЯ В РАСТВОРАХ НЕЭЛЕКТРОЛИТОВ. КРИОСКОПИЯ И ЭБУЛИОСКОПИЯ Экспериментальным путем (Рауль, 1887 г.) установлено, что давление пара растворителя над раствором ниже, чем над чистым растворителем и «относительное понижение давления насыщенного пара над раствором Δрравно мольной доле растворенного нелетучего вещества» (закон Рауля): где Закон Рауля справедлив для идеальных или бесконечно разбавленных растворов неэлектролитов (веществ, не распадающихся на ионы под действием полярных молекул растворителя). Из этого закона следует, что температура кипения разбавленного раствора, содержащего нелетучее растворенное вещество, выше, а температура замерзания соответственно ниже температуры кипения и замерзания чистого растворителя. Это следствие можно сформулировать так: «Повышение температуры кипения и понижение температуры замерзания раствора прямо пропорционально моляльной концентрации растворенного вещества»:
где
Измерение как разности температур замерзания
Моляльность Моляльность раствора раствора
отсюда
Для электролитов в закон Рауля вводится поправка — изотонический коэффициент i, который показывает, во сколько раз возрастает число частиц в растворе вследствие диссоциации электролита: Поэтому для растворов электролитов:
Изотонический коэффициент связан со степенью диссоциации α, которая представляет собой отношение числа продиссоциировавших молекул N к первоначальному числу N0:
Очевидно, что число продиссоциировавших молекул N будет равно N0α, а величина [N0 - N0α = N0 (I — α)] определит число непродиссоциировавших молекул. Общее же число частиц в растворе будет равно [ Изотонический коэффициент, таким образом, представляет собой отношение
По этому уравнению можно рассчитать степень диссоциации электролита, зная изотонический коэффициент, который в свою очередь определяется экспериментально как отношение полученного опытным путем значения свойства раствора, например следовательно:
Водородный показатель (рН) Вода является очень слабым электролитом. Её диссоциация – пример самоионизации, протекающей по уравнению Н20 + Н20 где Н3O+ — ион гидроксония, представляющий собой гидратированный ион водорода (Н20 + H+ = Н3O+). Константа диссоциации воды согласно закону действия масс:
Это произведение
В нейтральном растворе концентрации [Н3O+] и [ОН-] равны: [Н3O+] = [ОН-] =10-7моль/л. (2.5) Концентрация ионов гидроксония [Н3O+] определяет кислотность растворов. Если [Н3O+] > 1O-7 моль/л, то раствор является кислым, если [Н3O+] < 1O-7 моль/л — щелочным. Для более удобного выражения кислотности раствора было введено понятие рH, где р — оператор, тождественно равный отрицательному десятичному логарифму р ≡ -lg. Таким образом, величина рН представляет собой отрицательный десятичный логарифм числа, определяющего концентрацию ионов гидроксония [Н3O+] или упрощенно – ионов водорода [Н+]: pH = -lg[Н3O+]; pH = -lg[Н+]. (2.6) Оператор р применяется и для других малых величин, например pOH = -lg[OН+,, а р КW = pH + pOH = 14. (2.7)
pH нейтральных сред равен 7, кислых – меньше 7, щелочных – больше 7. Гидролиз солей. Ионообменная хроматография Хроматография представляет собой метод разделения и анализа смесей, который основан на различном распределении ее компонентов между неподвижной и подвижной фазами. Когда смесь веществ проходит через хроматографическую колонку, заполненную сорбентом (это мелкораздробленная неподвижная фаза), в ней происходят динамические про
В последнем случае сорбент представляет собой высокомолекулярные органические вещества (синтетические смолы), имеющие группы, содержащие либо подвижный ион водорода, либо подвижный ион гидроксила. В первом случае это могут быть сульфогруппы – S03Н, карбоксильные группы – СООН и др., Во втором – анионы ОН-, амминные группы NH2-, NH2- и др. Такие сорбенты называются ионитами. Иониты, обменивающиеся с раствором катионами, называются катионитами, а обменивающиеся анионами — анионитами. В общем виде реакции химического взаимодействия между ионитами и раствором можно представить следующим образом: ● на катионите m∙R-H + Mem+ ↔Rm-Me + m∙H+; (2.38) ● на анионите n∙R-OH + An-↔Rn-A + n∙OH-. (2.39) Обмен катионов и анионов между ионитами и раствором происходит в количествах, равных или пропорциональных химическим эквивалентам. Химическая реакция обмена ионов является обратимым процессом. Метод ионообменной хроматографии применяется, в частности, для очистки воды от различных примесей, причем о количестве последних, поглощенных сорбентом, можно судить по изменению электропроводности раствора. Так, при прохождении жидкости через катионит за счет ионного обмена в растворе появляются наиболее подвижные катионы – ионы водорода, вследствие чего удельная электропроводность раствора увеличивается. При пропускании раствора через анионит освобождающиеся ионы ОН- взаимодействуют с ионами водорода и образуют малодиссоциируемое соединение – воду Н20. Электропроводность раствора при этом уменьшается. Степень очистки раствора можно контролировать и по изменению его кислотности (рН). Вода, очищенная от положительно и отрицательно заряженных ионов методом ионообменной адсорбции, называется деионизированной.
2.6. Коллоидные растворы Коллоидными растворами называются мелкодисперсные системы, в которых существует граница раздела фаз между растворителем и растворенным веществом, частицы которого независимо друг от друга перемещаются путем броуновского движения. Радиус частиц, входящих в состав таких растворов, находится в пределах 10-7 м > r >10-9 м. Таким образом, коллоидные частицы нельзя увидеть в оптическом микроскопе, разрешающая способность которого ниже этих пределов. Коллоидные системы относятся к гетерогенным системам, которые содержат две фазы, например твердую и жидкую, равномерно распределенные друг в друге. Они широко распространены в природе, используются во многих производствах. К коллоидным растворам принадлежат многие естественные продукты, такие как молоко, кровь, яичный белок – это жидкие коллоидные системы. Твердые коллоидные системы представляют собой минералы (опалы, яшмы, агаты и др.). Атмосферный туман, вулканический дым – газообразные коллоидные системы.
Жидкие коллоидные системы называются золями. Они делятся на лиофобные (гидрофобные) и лиофильные (гидрофильные). Коллоидные растворы, в которых практически отсутствует взаимодействие между молекулами растворенного вещества и растворителя, называются лиофобными. Примером таких растворов служат коллоидные растворы металлов, раствор серы в воде. Дисперсные системы, для которых характерно интенсивное взаимодействие дисперсной среды с поверхностью дисперсной фазы, называются лиофильными системами. Частицы растворенного вещества (дисперсной фазы) при этом окружены молекулами растворителя (дисперсионной среды), образующими сольватную или гидратную оболочку. Лиофильные системы образуются самопроизвольно, например при диспергировании мыла, глины, полимеров в растворителе под действием теплового движения. Смачиваемость поверхности какого-либо вещества возможна лишь при использовании растворов, обладающих лиофильными свойствами по отношению к природе поверхности данного вещества. Учитывая тот факт, что протекание любого гетерогенного процесса (травления, осаждения покрытий и др.) начинается с процесса смачивания, очевидна необходимость тщательного подбора реагирующих веществ для подготовки поверхности. По Н.Д. Пескову, следует различать два вида устойчивости коллоидных систем: кинетическую (седиментационную) и агрегативную. Кинетическая устойчивость характеризует устойчивость к оседанию частиц при их тепловом (броуновском) движении в растворе. Чем больше масса частиц, тем меньше их кинетическая устойчивость. Агрегативная устойчивость характеризует устойчивость системы против слипания частиц при их столкновении друг с другом. Она зависит от степени сольватации (гидратации) растворенных частиц, от величины энтропии системы, характеризующей степень ее хаотичности, и от кинетической энергии частиц. Если система теряет агрегативную устойчивость, то коллоидный раствор коагулирует, т.е. происходит слипание частиц с образованием более крупных агрегатов и их осаждение или всплытие на поверхность. При этом происходит и потеря кинетической устойчивости системы. Элементарная частица коллоидных систем называется мицеллой. Она состоит из ядра, нерастворимого в дисперсионной среде, адсорбционного и диффузного слоев. Ядро представляет собой агрегат многих молекул, которые могут иметь различный состав, например [Fе(ОН)з]n, [AgJ] n и т.д., где n – число молекул, входящих в состав агрегата, и адсорбционного слоя, который состоит из ионов, преимущественно общих с ионами, составляющими ядро (правило Пескова – Фаянса). Ядро коллоидной частицы {[Fе(ОН)з]n ∙m Fe+3}+3m заряжено положительно, благодаря адсорбции потенциалопределяющих ионов Fe+3 на поверхности ядра. Так как в целом мицелла электронейтральна, этот положительный заряд уравновешивается равным отрицательным зарядом противоионов, например ионов С1-. Силы взаимодействия в этом случае имеют электростатическую и адсорбционную природу. Те ионы С1-, которые прочно связаны с ядром и входят в состав адсорбционной зоны, называются противоионами адсорбционного слоя и составляют вместе сядром гранулу (частицу). Те же ионы, которые из-за собственного теплового движения распределяются диффузно, называются противоионами диффузного слоя. Разделение окружающих ядро противоионов на две части – одну, находящуюся внутри границы скольжения и движущуюся совместно с ним, и вторую, «отрывающуюся» под действием внешнего поля от поверхности частицы, – позволяет записывать своебразные «химические формулы», отражающие строение мицелл коллоидных систем. Для гидрозоля гидроксида железа строение мицеллы имеет следующий вид:
{[Fе(ОН)з]n∙mFe+3 ∙ 3(m-x)Cl-}3x+ · 3xCl- (2.40) ядро адсорбционный диффузный слой слой [ ____________________гранула________ ]
[___________________ мицелла _______________]
Рис. 2.1. Изменение потенциала в двойном электрическом слое
Из структуры мицеллы видно, что на межфазной границе возникает двойной электрический слой, который обладает определенным потенциалом. Внешний электрический слой состоит из двух частей. Первую составляют противоионы, которые находятся вблизи ядра и удерживаются около него силами электростатического притяжения (толщину этого слоя обозначим буквой «а») (рис. 2.1); вторую часть составляют противоионы, диффузно распределенные по объему (слой «в»). В слое «а» изменение потенциала носит линейный характер (по аналогии с конденсатором), а в слое «в» (диффузном) – экспоненциальный. Тот потенциал, который возникает на границе скольжения фаз относительно друг друга (между гранулой и диффузным слоем), называется электрокинетическим (или ξ- потенциалом). Чем больше концентрация электролита и заряд его ионов, тем меньше толщина слоя «а» и величина ξ -потенциала. Это обусловлено тем, что с ростом числа ионов, определяющих заряд ядра, большее число противоионов переходит из диффузной части двойного электрического слоя в адсорбционную. Если приложить к коллоидному раствору внешнюю разность потенциалов, то в них наблюдается либо направленное перемещение частиц к электродам согласно знакам их зарядов (явление электрофореза), либо направленное перемещение одной жидкой фазы относительно другой (явление электроосмоса). Устойчивость коллоидных систем может быть нарушена путем добавления к ним сильных электролитов. Коагулирующее действие электролитов на коллоидные системы характеризуется порогом коагуляции, т.е. наименьшей концентрацией электролита, вызывающей коагуляцию. При добавлении электролита к лиофобному коллоидному раствору за счет вытеснения противоионов из диффузной части мицеллы в адсорбционную толщина двойного электрического слоя уменьшается. В случае лиофильных коллоидных растворов введение электролита способствует разрушению сольватной оболочки. Силы притяжения между частицами начинают возрастать, и наступает быстрая коагуляция. Высокомолекулярные соединения (ВМС) и лиофильные коллоиды являются стабилизаторами по отношению к лиофобным золям. Так, если прибавить к коллоидному раствору соли серебра, например AgI, небольшое количество желатина или белка, и восстановить серебро до образования золя, то степень дисперсности коллоидного серебра в этих условиях его получения оказывается более высокой и золь менее подвержен влияниям факторов, вызывающих коагуляцию. Вследствие защитного действия, которое в подобных случаях оказывают лиофильные коллоиды, повышая стабильность золей, их называют защитными коллоидами. То минимальное количество высокомолекулярного вещества, которое необходимо для защиты 10 мл коллоидного раствора от коагуляции, называется защитным числом: где С – процентное содержание защищающего вещества; V – объем защищающего вещества; W – объем коллоидного раствора. Раздел II. ЭЛЕКТРОХИМИЯ Глава 4. ЭЛЕКТРОЛИЗ Электролиз – это окислительно-восстановительные процессы, которые происходят на электродах при прохождении электрического тока через раствор электролита или его расплав. В этом случае электрическая энергия источника тока переходит в химическую энергию. Электролиз применяют главным образом при нанесении защитных покрытий на металлические изделия, при получении различных Для осуществления процесса электролиза расплавов или растворов электролитов используется электрохимическая ячейка, состоящая из двух электродов – катода и анода, присоединенных к источнику постоянного тока и соответствующего электролита. По возможности участия в окислительно-восстановительных реакциях, определяющих электрохимический процесс, аноды подразделяются на нерастворимые (не участвующие в реакции, чаще всего платиновые, угольные и другие инертные электроды) и растворимые, принимающие участие в электродной реакции и при этом растворяющиеся (в этом случае металл анода подбирается, исходя из целей и задач самого процесса электролиза). Общие правила разряда ионов при электролизе. В первую очередь на электродах будет реагировать тот ион, для разряда которого нужно приложить меньшее значение (по абсолютной величине) потенциала. Так как на катоде идет процесс восстановления, т.е. присоединения отрицательно заряженных электронов, то первоначально на нем будет протекать тот электродный процесс, который характеризуется более положительным значением потенциала, а на аноде − электродный процесс, характеризующийся более отрицательным значением потенциала, так как там идет окисление, сопровождающееся отдачей электронов. Очередность разряда ионов металлов определяется положением последних в ряду напряжений. Группа металлов «высокой» активности: Li+, Rb+, K+, Ca2+, Na+, La+, Mg2+, Be2+, Al3+ При электролизе водных растворов «активных» металлов на катоде происходит выделение водорода из воды, а ионы металла остаются в растворе в первоначальном виде, как и до электролиза, так как стандартный электродный потенциал реакции восстановления водорода из воды 2 Н2О + 2 е ↔ Н2 + 2 ОН- составляет величину В группу металлов «средней» активности входят: Mn2+, Zn2+, Fe2+, Cd2+, Co2+, Ni2+, Sn2+,Pb2+, Ge2+.. Их стандартные потенциалы определяются пределами от -1,53 В для Zn2+ до -0.01 В для Ge2+.. При электролизе водных растворов металлов «средней» активности на катоде происходят две реакции − восстановление как металла, так и водорода из воды. Группа «неактивных» металлов, стоящих в ряду активности после водорода: Bi2+, Cu2+, Hg2+, Ag+, Au3+, Au+, Стандартные потенциалы «неактивных» металлов всегда имеют положительное значение и охватывают область потенциалов от +0,22 В для Bi2+ до +1,68 В для Au+. Водороду (т.е. водородному электроду) Pt, H2(Г)│2H+ условно приписывается нулевое значение стандартного потенциала. При электролизе водных растворов «благородных» металлов, т.е. имеющих положительное значение стандартного электродного потенциала, на катоде происходит восстановление только металла. Ниже приведен ряд напряжений металлов с указанием ионов, которые образуются при их окислении:
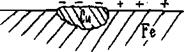
Li+, Rb+, K+, Ca2+, Na+, La+, Mg2+, Be2+, Аl3+, Mn2+, Zn2+, Fe2+, Cd2+, Co2+, Ni2+, Sn2+, Pb2+, Ge2+., H+ Bi2+, Cu2+, Hg2+, Ag+, Au3+, Au+ Явление перенапряжения, имеющее место при протекании тока через электролизер, приводит к тому, что водород на катоде выделяется при потенциале гораздо более электроотрицательном, чем равновесный потенциал, отвечающий рН данного раствора. Перенапряжение выделения кислорода при его образовании на аноде по схемам ● в щелочном или нейтральном растворе 4ОН— -4е → О2+2Н2О (рН >7); (4.1) ● в кислом растворе 2Н2О -4е → О2 +4Н+ (рН <7) (4.2) заключается в смещении его потенциала в сторону положительных значений от равновесного потенциала. Перенапряжение в общем случае представляет собой сдвиг потенциала электрода от данного равновесного значения, который вызывается замедленностью стадии разряда электрохимической реакции. Рассмотрим конкретно процесс электролиза раствора и расплава соли NaCl. При электролизе расплава NaCl, в котором существует только один вид катионов и один вид анионов, образующихся при термической диссоциации NaCl по схеме NaCl ↔ Na+ + Cl- протекают следующие процессы: на отрицательно заряженном катоде К(-) происходит восстановление ионов натрия и образование чистого металла, а на положительно заряженном аноде А(+) − окисление ионов хлора с образованием газообразного продукта: (-)К: Na+ + e → Nao (4.3) +)A: С1- -e → С1° (4.4) С1° + С1°→ С12. Если же проводить электролиз водного раствора NaС1, то катодный процесс в этом случае будет иной. Это связано c тем, что в водном растворе электролизу будут подвергаться молекулы воды, как обладающие более положительным значением электродного потенциала, и для того чтобы произошло восстановление воды на катоде с образованием водорода в случае щелочных и нейтральных растворов по схеме 2H2O + 2e = H2 + 2 OH- (4.5) надо приложить потенциал, равный Ео = -0,83 В, а для восстановления ионов Na+ требуется создать потенциал, равный Ео = -2,7 В, поэтому как только потенциал катода станет равным -0,83В, на катоде начнется выделение водорода из воды. Это приводит к тому, что получать активные металлы с помощью электролиза возможно только из расплавов их соединений, когда отсутствует конкурирующая реакция восстановления воды. На аноде в первую очередь разряжается ион, который имеет менее положительный потенциал. Для окисления на аноде воды с выделением кислорода по реакции 2H2O - 4e → О2 + 4H+ следует приложить потенциал Е0 = 1,23В, а для окисления ионов хлора 2С1--2e →С12° требуется +1,36В. Однако в водном растворе NaС1 на аноде происходит выделение хлора, из-за сдвига потенциала в связи с процессом перенапряжения выделения кислорода. Таким образом, на аноде при электролизе водных растворов солей, имеющих простые анионы типа S2-, J-, Br-, Cl-, происходит их окисление с выделением свободных неметаллов. В случае же электролиза соединений, содержащих кислородсодержащие анионы типа PO43-, SO42-, NO3- и ион фтора F-, происходит окисление воды с образованием кислорода, так как для окисления последних требуется потенциал более 2 В. При электролизе водного раствора сульфата меди CuSO4 с нерастворимым анодом (например, угольным) протекают следующие реакции: на катоде происходит восстановление металла (-) К: Cu2+ + 2е → Cuо, на аноде окисление воды с выделением кислорода (+)А: 2H2O - 4e → О2 + 4H+. При электролизе водных растворов солей неактивных металлов и металлов «средней» активности при наличии растворимого анода возможно протекание сопряженных реакций на аноде и катоде. Так, для сульфата никеля, молекулы которого распадаются в воде на ионы Ni2+ и SO42-, возможно протекание электролиза по следующей схеме: ● на катоде (-)К: Ni2+ + 2е → Ni0 2H2O + 2e → H2 + 2 OH- (4.6) ● на аноде (+)А: Ni0 - 2е → Ni2+ 2H2O - 4e → О2 + 4H+ . (4.7) В случае же нерастворимого анода электролиз может быть представлен электродными реакциями: ● на катоде (-)К: Ni2+ + 2е → Ni0 ● на аноде (+)А: 2H2O - 4e → О2 + 4H+. В соответствии с законами Фарадея количество вещества m, претерпевшего химические превращения на электроде при прохождении тока через раствор или расплав электролита, прямо пропорционально количеству электричества Q и эквивалентной массе данного вещества Э: где F – число Фарадея ≈ 96500 кл/г-экв. Ели на электродах протекают конкурирующие реакции, то на выделение целевого продукта расходуется только часть количества электричества, протекающего через электрод, и тогда говорят о выходе по току целевого продукта. При электрохимическом получении металлов практически всегда часть тока расходуется на побочные процессы, такие, как выделение водорода и др., поэтому количество металла, выделившееся на электродах, будет меньше, чем рассчитанное теоретически по законам Фарадея. Отношение количества вещества, получаемого при электролизе, к его теоретическому значению называется выходом по току η: ОТ КОРРОЗИИ Коррозия металлов − самопроизвольный процесс разрушения (окисления) металла за счет его взаимодействия с окислителем окружающей среды. Основные механизмы протекания коррозионных процессов − электрохимический и химический. Критерием протекания коррозии по одному из перечисленных выше механизмов является состав (природа) среды. Химической коррозии подвергаются металлы, находящиеся в средах, не проводящих электрический ток, например в сухих газах в присутствии окислителей О2, СО2, Сl2, J2, H2S и других (газовая коррозия), в неводных растворах (раствор йода в бензоле, сернистая нефть) или в агрессивных средах, не содержащих воды (концентрированные растворы кислот, щелочей и т.д.). Газовой коррозии подвержены сопла ракет, двигатели внутреннего сгорания и т.п. В этом случае разрушение металла происходит за счет его непосредственного взаимодействия с окислителем. Более распространенной является электрохимическая коррозия металлов, так как в основном изделия из металлов и сплавов эксплуатируются в атмосферных условиях, в которых присутствует вода в большем или меньшем количестве в зависимости от природы среды: почва, влажный воздух, полное погружение изделия в электролит и т.п. Таким образом, электрохимической коррозии (критерий – токопроводящая среда) подвержены те металлические изделия, которые находятся в растворах электролитов или во влажном воздухе. Это электрохимический процесс, так как на поверхности металла имеются участки с различным значением электродного потенциала и образуются так называемые короткозамкнутые микрогальванопары и вследствие этого электрический ток. Электрохимическая коррозия – многостадийный процесс, основными стадиями которого являются: окисление металла на анодных участках корродирующего металла и восстановление окислителя из окружающей среды − на катодных. Рассмотрим в качестве примера коррозию железа в растворе НСl.
Рис. 5.1. Коррозия железа в кислой среде
|
|||||||||||
|
|
Последнее изменение этой страницы: 2021-03-10; просмотров: 195; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.149.242.253 (0.152 с.) |
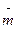 — число молей растворенного вещества в 1000 г растворителя [г-моль/кг растворителя];
— число молей растворенного вещества в 1000 г растворителя [г-моль/кг растворителя];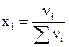 .
.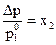 , (1.1)
, (1.1)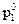 — давление насыщенного пара чистого растворителя;
— давление насыщенного пара чистого растворителя; 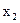 — мольная доля растворенного нелетучего вещества.
— мольная доля растворенного нелетучего вещества.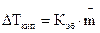 ;
; 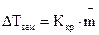 ,
,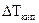 – повышение температуры кипения раствора;
– повышение температуры кипения раствора; 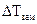 – понижение температуры замерзания раствора;
– понижение температуры замерзания раствора; 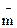 — моляльность раствора;
— моляльность раствора; 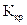 – криоскопическая постоянная;
– криоскопическая постоянная; 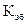 –
–  эбуллиоскопическая постоянная.
эбуллиоскопическая постоянная.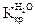 = 1,86о,
= 1,86о, 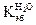 = 0,52о для бензола
= 0,52о для бензола 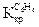 = 4,90о,
= 4,90о, 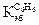 = 2,65о.
= 2,65о.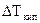 (эбулиоскопия) раствора и чистого растворителя дает способ экспериментального определения молярной массы неэлектролита. Для этого навеску в граммаx
(эбулиоскопия) раствора и чистого растворителя дает способ экспериментального определения молярной массы неэлектролита. Для этого навеску в граммаx 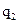 вещества с неизвестной молярной массой М2, содержащей
вещества с неизвестной молярной массой М2, содержащей 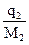 молей растворенного вещества, растворяют в известном количестве растворителя
молей растворенного вещества, растворяют в известном количестве растворителя 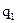 и измеряют повышение температуры кипения или понижение температуры замерзания:
и измеряют повышение температуры кипения или понижение температуры замерзания: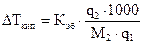
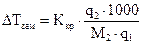 , (1.3)
, (1.3) 
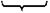
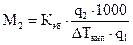 , или
, или 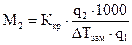 . (1.4)
. (1.4)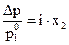 . (1.5)
. (1.5)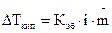
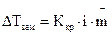 . (1.6)
. (1.6)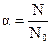 . (1.7)
. (1.7)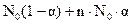 ], где n – суммарное число ионов, на которое распадается одна молекула электролита.
], где n – суммарное число ионов, на которое распадается одна молекула электролита.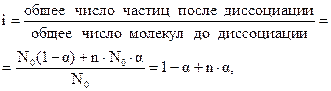
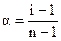 . (1.7 а)
. (1.7 а) к теоретическому (расчетному) значению:
к теоретическому (расчетному) значению: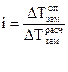 , (1.7 б)
, (1.7 б)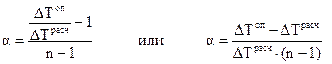 . (1.8)
. (1.8) Н3O+ + ОН-,
Н3O+ + ОН-,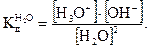 (2.4)
(2.4)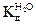 при 25оС равна 3,24∙I0-18. Так как степень диссоциации молекул воды очень мала, то концентрацию молекул воды можно считать величиной постоянной и равной [H2O] = 1000/18 = 55,55 моль/л и произведение двух постоянных величин тоже величина постоянная, равная:
при 25оС равна 3,24∙I0-18. Так как степень диссоциации молекул воды очень мала, то концентрацию молекул воды можно считать величиной постоянной и равной [H2O] = 1000/18 = 55,55 моль/л и произведение двух постоянных величин тоже величина постоянная, равная: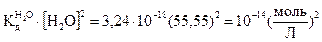 .
.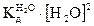 = [Н3O+][ОН-] = IO-14 = КW (моль/л)2 является постоянным для всех водных растворов. Константа КW называется ионным произведением воды, она зависит только от температуры.
= [Н3O+][ОН-] = IO-14 = КW (моль/л)2 является постоянным для всех водных растворов. Константа КW называется ионным произведением воды, она зависит только от температуры.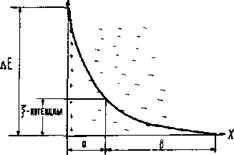
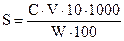 , (2.41)
, (2.41)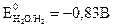 , а стандартные потенциалы «активных» металлов колеблются в пределах от -3,045 В для Li+ до -1,670 В для Al3+.
, а стандартные потенциалы «активных» металлов колеблются в пределах от -3,045 В для Li+ до -1,670 В для Al3+.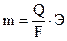 , (4.8)
, (4.8)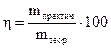 [%]. (4.9)
[%]. (4.9)