Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Микроаномалии развития, определение, цель выявления, особенности клинико-морфологического осмотра врачом генетика.Содержание книги
Поиск на нашем сайте
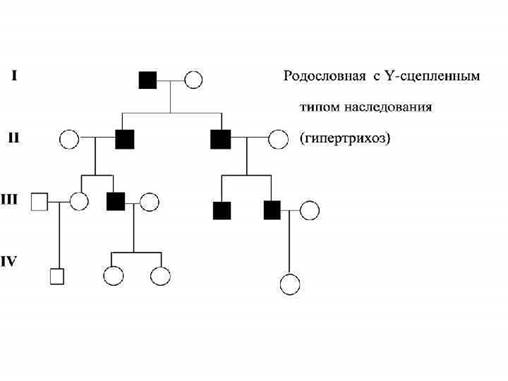
Микроаномалии — это небольшие морфологические изменения органов, несколько выходящие за пределы нормального строения, но не нарушающие их функции. Микроаномалии, выявляемые у больных с врожденными и наследственными заболеваниями или синдромами, часто не ограничены одним органом или тканью. Они обнаруживаются в различных органах, развивающихся из разных зародышевых листков. Большинство микроаномалийвыявляются в периоде новорожденности, однако некоторые микроаномалии не являются статичными, с ростом и развитием ребенка они могут изменяться и даже исчезать. Исследования показывают, что наличие 1-3 микроаномалий развития встречается у здоровых лиц. Для выработки единого взгляда на MAP следует обращать внимание на два положения: количество аномалий и их качественные характеристики. Ход постановки ДИАГНОЗА наследственного заболевания поэтапный: Первый этап - общее клиническое обследование больного, включающее анализ наследственного анамнеза, синдромологический анализ признаков болезни, выявление микроаномалий; Второй этап - необходим при подозрении на конкретную наследственную болезнь (по итогам первого). С этой целью привлекаются основные методы исследования медицинской генетики. 20. Виды генных мутаций. Нонсенс, миссенс, сдвиг рамки считывания, сплайнсинговые. Механизмы. По характеру изменений в составе гена различают следующие типы мутаций: Делеции — утрата сегмента ДНК размером от одного нуклеотида до гена. Дупликации — удвоение или повторное дублирование сегмента ДНК от одного нуклеотида до целых генов. Инверсии — поворот на 180° сегмента ДНК размером от двух нуклеотидов до фрагмента, включающего несколько генов. Инсерции — вставка фрагментов ДНК размером от одного нуклеотида до целого гена. Трансверсии — замена пуринового основания на пиримидиновое или наоборот в одном из кодонов. Транзиции — замена одного пуринового основания на другое пуриновое или одного пиримидинового на другое в структуре кодона.
Генные мутации происходят на молекулярном уровне и затрагивают, как правило, один или несколько нуклеотидов внутри отдельного гена. Этот тип мутаций можно разделить на две большие группы. Первую из них обуславливает сдвиг рамки считывания. Ко второй группе относят генные мутации, связанные с заменой пар оснований. Последние составляют не более 20% спонтанных мутаций, остальные 80% мутаций происходят в результате различных делеций и вставок. Мутации со сдвигом рамки считывания представляют собой вставки или выпадения одной или нескольких пар нуклеотидов. В зависимости от места нарушения изменяется то или иное количество кодонов. Соответственно в белке могут появиться дополнительные аминокислоты или измениться их последовательность. Большая часть мутаций этого типа обнаружена в молекулах ДНК, состоящих из одинаковых оснований. Типы замены оснований: 1. Транзиции заключаются в замене одного пуринового на пуриновое основание или одного пиримидинового на пиримидиновое основание 2. Трансверсии, при которых пуриновое основание меняется на пиримидиновое или наоборот. Значимость генных мутаций для жизнеспособности организма неодинакова. Различные изменения в нуклеотидной последовательности ДНК по-разному проявляются в фенотипе. Миссенс-мутации связаны с заменой нуклеотидов в кодирующей части гена. Фенотипически проявляется в виде замены аминокислоты в белке. В зависимости от природы аминокислот и функциональной значимости нарушенного участка, наблюдается полная или частичная потеря функциональной активности белка. Сплайсинговые мутации затрагивают сайты на стыке экзонов и интронов и сопровождаются либо вырезанием экзона и образованием делегированного белка, либо вырезанием интронной области и трансляцией бессмысленного измененного белка. Как правило, такие мутации обусловливают тяжелое течение болезни. Нонсенс-мутация — замена нуклеотида в кодирующей части гена — приводит к образованию кодона-терминатора (стоп-кодона) и прекращению трансляции. Генные болезни. Общие понятия. Классификация. Примеры. Генные болезни - это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена Виды генных мутаций: · замена пар нуклеотидов (миссенс- мутации и нонсенс- мутации) · выпадения (делеция) одной или нескольких пар нуклеотидов · вставка нуклеотида (инсерция) · экспансия тринуклеотидных повторов · нарушение сплайсинга Результатом миссенс- мутации являются: · серповидно-клеточная анемия (замена глутаминовой кислоты в молекуле HbA на валин в 6 положение в молекулах HbS) · различия по инсулину: треонин в 30 положение b-цепи- инсулин человека; аланин- инсулин свиньи; серин- инсулин кролика; a-цепь у всех одинакова · a-b- талассемия · иммуноглобулинjпатия · муковисцидоз Dставки (инсерции) вызывают аналогичные изменения рамки считывания только в другую сторону пр.миодистрофияДюшенна и Беккера, муковисцидоз Экспансия тринуклеотидных повторов -в норме число одинаковых триплетов в нити ДНК постоянно, при экспансии тринуклеотидных повторов их чего может увеличиваться в несколько раз Пр. · хорея Гентингтона (в 4 хромосоме число триплетов ЦАГ увеличивается в 3 раза) · миотическая дистрофия (в 19 хромосоме число повторов ЦТГ увеличивается в 400 раза и более) · синдром fra-x- умственная отсталость с ломкой x-хромосомой (триплет ЦГГ повторяется от 200 до 2000 раз (в норме 6-51 раз)) · спинно- церебральная атаксия в 6 хромосоме триплет ЦАГ повторяются до 80 раз (в норме 25-36) Мутация в одной из клеток на ранних стадиях дробления приводит к возникновению мозаичныхформ (обнаружены при миопатии Дюшенна) Если мутации в клетках половых органов- мозаицизм гонад Частота мозаицизм у родителей составляет 5-15% случаев в доминантных и Х- сцепленных рецессивных болезней: · несовершенный остеогенез · синдром Элерса-Данло · гемофилия 22. Клинико-генеалогический метод. Типы наследования. Клинические примеры. Клинико-генеалогический метод позволяет: · выявить наследственный характер заболевания · установить тип наследования болезни · установить зиготность членов членов родословной · определите вероятность появления признаков в потомстве · дать обоснованный прогноз для потомства в семье, где есть носитель патологического гена · изучить гетерозиготность наследственных болезней Показания: · наличие у больного и его родственников моногенного заболевания · заболевания с наследственной предрасположенностью · семейный характер заболевания · непереносимость некоторых пищевых продуктах · извращенная реакция на действие лекарств · аллергические заболевания в семье · кровное родство родителей больного ребенка · отягощенный акушерский анамнез · врождённые пороки развития Клинико-генеалогический метод лежит в основе медико-генетического консультирования и включает 3 этапа: 1 этап- сбор клинического анамнеза и клиническое обследование 2 этап- составление родословной 3 этап- генетический анализ родословной Анамнез настоящего заболевания (начало, характер, течения) · Рецидивирующая, хроническая, длительно не поддающаяся лечению течения, особенно в детском возрасте (хроническая пневмония при муковисцидозе, расстройство кишечника при целиакии, дисахаридазная недостаточность) · стойки изменения в моче (протеинурия, гематурия)- синдром Альпорта или семейная гематурия · стойкая гипотрофия (муковисцидоз, целиакия, дисахаридазная недостаточность) · прогрессирующее, не поддающиеся лечению судорожные приступы · наличие у больного редких, специфических симптомов или их сочетаний · вовлечение в патологический процесс многих органов и систем. Ообенно распространённый характер поражения наблюдается при хромосомной патологии Анамнез жизни: · течение беременности и родов, периода новорождённости, вскармливание, физическое и нервно-психическое развитие, перенесённые заболевания, данные акушерского анамнеза · сведения о сибсах · сведения о родителей · национальность (болезнь Гоше, болезнь Ниманна-Пика у евреев, некоторые формы наследственных аниме у негров) · места жительства семьи (влияние эндемического фактора) · места жительства предков (выявление кровно родственных браков) · профессия (профессиональные вредности, приём лекарственных препаратов во время беременности- возможность тератогенного действия на плод) Объективное обследование пробанда: · детальный осмотр и его и родственников · антропометрия · описание фенотипических проявлений заболевания Синдром Элерса-Данло, нейрофиброматоз Патология мышечной ткани: · характерно во многих наследственных синдромов · аплазии ряда мышцы верхних конечностей (при синдроме Эдвардса) Патология костной системы: · деформация грудной клетки, черепа и позвоночника, слегка согнутые конечности, широкие · хрупкие кости, множественное спонтанные переломы- несовершенный остеогенез · длинные, тонкие паучьи пальцы, изменённое грудная клетка- патогномоничный признак болезни Марфана Антропометрические исследования: · рост · массы тела · телосложения · длина конечностей · окружность груди и головы · соотношения сагиттального энтерального размеров черепом Высокий рост, длины конечности, арахнодактилия указывают на синдром марфана Укорочении конечностей по сравнению с туловищем, запавшая переносица- ахондроплазия. Микроцефалия- симптомы их наследственных болезней 23. Особенности аутосомно-доминантного типа наследования. Составить родословную. Аутосомно-доминантный тип наследования характеризуется: · патологический признак встречается в каждом поколении и проявляется у гетерозигот; соотношение здоровых и больных чипсов приближается 1: 1 · чаще неполная пенетрантность признака (пр. пенетрантность для ретинобластомы примерно 90%) · различная выраженность клинических проявлений не только между разными семьями, но и внутри каждой семьи (пр.нейрофиброматоз у одних членов семьи- генерализованная форма, а у других- отдельные кожные проявления; синдром марфана) · клинические проявления могут развиваться спустя несколько лет после рождения, а сроки проявления у разных членов семьи разные (пр. хорея Гентингтона, миопическая дистрофия, которое может иметь начала от пренатального периода до 50 60 лет) Близорукость Аутосомно-рецессивный тип наследования характеризуется: · риск рождения больного ребенка 25%, если родители здоровы и гетерозиготное · если родители гомозиготны, то все сибсы должны быть больными (пр. альбинизм, фенилкетонурия) · браке гетерозигот с гомозиготами дают менделеевское расщепление больных и здоровых 1: 1 Это главным образом кровнородственные браки По аутосомно рецессивному признаку наследуется в основном энзимопатии, многие из них с известными биохимическим дефектом Особености Y сцепленного типа наследования. Составить родословную. Примеры заболеваний. 1. Гены расположены в Y хромосоме 2. Болезнь передается только от отца 3. Заболевание наследуется только мальчиками 4. Признак наблюдается в каждом поколение по отцовской линии 5. Примеры: гипертрихоз ушной раковины, перепонка между пальцами.
Особенности Х сцепленного доминантного типа наследования. Родословная. Примеры. 1. если болен отец, то все его дочери- больные а все сыновья, здоровые, 2. больными будут дети только в том случае, если болен один из родителей 3. у здоровых родителеи все дети будут здоровы 4. заболевание прослеживается в каждом поколении 5. если мать больна, то вероятность рождения больного ребенка равна 50 % независимо от пола; 6. болеют как мужчины,так и женщины, но в целом больных женщин в семье в 2 раза больше, чем больных мужчин. 7. Например, Витамин - Д-резистентный рахит
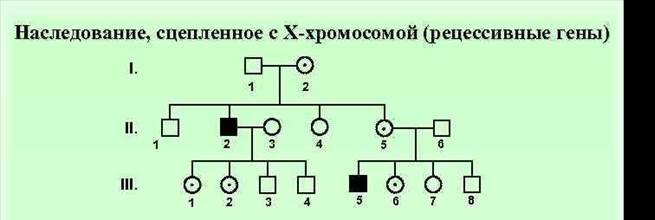
Особенности Х сцепленного рецессивного типа наследования. Родословная. Примеры. 1. преимущественно поражаются мужчины 2. признак проявляется фенотипически не в каждом поколении, поэтому от брака больного мужчины и здоровой женщины все дети здоровы 3. заболевание наблюдается у мужчин - родственников пробанда по материнской линии. 4. сын никогда не наследует болезни отца; 5. при браке между здоровым мужчиной и гетерозиготной женщиной вероятность рождения больного ребенка составляет 25 % для мальчиков и 0 % для девочек 6. Так наследуется гемофилия, дальтонизм,мужское бесплодие
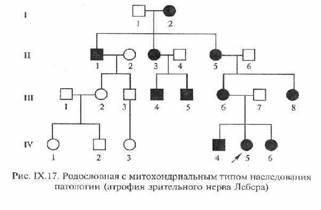
29. Передача генетической информации через цитоплазму получила название цитоплазматической (внеядерной, нехромосомной) наследственности. Поскольку наследственная информация передается по материнской линии через цитоплазму яйцеклетки, ее называют также материнской наследственностью. Непосредственное влияние материнского организма на развитие зародыша часто приводит к большему сходству потомства с матерью, поскольку условия эмбрионального развития организма полностью зависят от матери.

30. Наследственные болезни обмена – группа моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки, что приводит к нарушению метаболизма клетки.
|
||||||||
|
|
Последнее изменение этой страницы: 2021-01-08; просмотров: 735; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.41 (0.009 с.) |

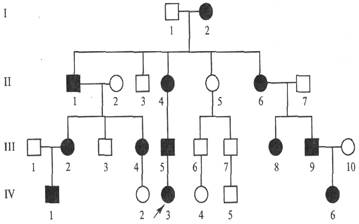 Родословная с Х-сцепленным доминантным типом наследования заболевания (витамин-Д-резистентный рахит)
Родословная с Х-сцепленным доминантным типом наследования заболевания (витамин-Д-резистентный рахит)